
08 Nov
FDA gives full recognition to AAMI cybersecurity guidance document
FDA gives full recognition to AAMI cybersecurity guidance document
Introduction
The Association for the Advancement of Medical Instrumentation (AAMI) is celebrating a significant achievement as the U.S. Food and Drug Administration (FDA) grants full recognition to AAMI’s pioneering guidance document on medical device cybersecurity, ANSI/AAMI SW96:2023, Standard for medical device security – Security risk management for device manufacturers. This recognition from the FDA signifies a crucial milestone in ensuring the security and integrity of medical devices, ultimately contributing to better patient outcomes. In this blog post, we will delve into the key aspects of this groundbreaking cybersecurity guidance document, its implications for the medical device industry, and the critical role it plays in safeguarding healthcare technology.
AAMI: A Beacon of Expertise in Healthcare Technology
Before we explore the details of the ANSI/AAMI SW96:2023 standard, let’s take a moment to understand the Association for the Advancement of Medical Instrumentation (AAMI). Established in 1967, AAMI is a nonprofit organization comprising more than 10,000 healthcare technology professionals with a shared mission: to support the development, management, and use of safe and effective health technology in the healthcare community. AAMI is recognized as a primary source of consensus standards, both nationally and internationally, for the medical device industry. Additionally, AAMI provides practical information, support, and guidance to health technology and sterilization professionals.
The Evolution of Medical Device Cybersecurity Standards
The introduction of ANSI/AAMI SW96:2023 represents a significant advancement in the field of medical device cybersecurity. To appreciate the significance of this standard, let’s briefly review the landscape of cybersecurity standards in the medical device industry before its release.
Prior to SW96, several guides and standards addressed security risk management for medical devices, including AAMI TIR57 and TIR97, UL 2900-2 and UL 2900-2-1 for Network Connectable Components, IEC 81001-5-1 for Health software and health IT systems safety, and various guidance documents from regulatory authorities and international consortiums such as the FDA, MDCG, TGA, and IMDRF. While these documents provided valuable insights, SW96 represents a leap forward in terms of detail and comprehensiveness.
AAMI SW96: A Comprehensive Guide to Medical Device Cybersecurity
AAMI SW96 devotes a substantial 14 pages to describing a robust security risk management process for medical devices. This level of detail sets it apart from its predecessors and is on par with ISO 14971, a recognized standard for risk management in medical devices. The standard’s intent is clear: to specify requirements on how medical device manufacturers should manage security risk throughout a device’s lifecycle within the risk management framework defined by ISO 14971.
Key Priorities in AAMI SW96
- Risk Management Process in Design: SW96 outlines the classical steps of a risk management process, including identifying, assessing, determining risk controls, verifying their effectiveness, evaluating overall residual risk acceptability, and considering the risk-benefit balance.
- Risk Management Process in Post-Production: The standard emphasizes the importance of establishing an enterprise-wide process for managing security post-production. It calls for the creation of design features that facilitate production and post-production security management, along with integration into healthcare delivery organization (HDO) network security policies and technologies.
- Monitoring, Control, and Coordination: Effective risk management necessitates coordination with users, customers, and integrators. SW96 emphasizes the coordination of communications with HDOs for security risks and the importance of understanding and conveying security expectations from manufacturers to those deploying medical devices in user environments.
- Delivery of Patches and End-of-Life: The standard underlines the significance of change control and the design process in delivering security patches promptly. Manufacturers are also urged to anticipate safe and secure device decommissioning and end-of-life.
Detailed Requirements and Precision
AAMI SW96 goes beyond its predecessors in providing precise requirements for security risk management during production and post-production phases. It insists on the monitoring of risk control measures’ effectiveness during production and outlines detailed requirements for information collection, review, and subsequent actions.
Security Controls and Misuse
While SW96 offers extensive guidance on risk management processes, it remains vague on specific security controls. Instead, it points to relevant standards, technical reports, and security frameworks for these details. However, it does address the prioritization of security controls, emphasizing inherent security by design, manufacturing processes, threat mitigation measures, risk disclosure, and security training.
The standard also addresses the concept of reasonably foreseeable misuse, taking into account exploit scenarios by threat actors and failures of end-users to follow security best practices.
Benefit-Risk Analysis and Residual Risk Acceptability
AAMI SW96 introduces the possibility of conducting a benefit-risk analysis when security risks remain unacceptable despite practical risk controls. This provision is particularly relevant for legacy devices, where balancing direct medical benefits against overall security residual risk becomes crucial.
Supply Chain and Third-Party Devices
SW96 tackles supply chain management and third-party medical device security, mandating the documentation of supply chain management in the security risk management plan and third-party device security in the security risk management file.
End of Guaranteed Support (EOGS) and End of Support (EOS)
AAMI SW96 introduces the concepts of EOGS and EOS to guide the phased withdrawal of devices reaching their end of life. These concepts are elaborated upon in the IMDRF document on cybersecurity for legacy medical devices.
Conclusion: A Standard Setting the Bar
In conclusion, ANSI/AAMI SW96:2023 is poised to become the cornerstone for security risk management in the medical device industry. Its detailed and comprehensive approach ensures its applicability to manufacturers dealing with Software as Medical Devices (SaMD) and Software in Medical Devices (SiMD). We can anticipate that this standard will gain recognition from regulatory bodies, including the FDA.
Moreover, AAMI SW96’s relevance extends beyond national borders, making it a potential candidate for international standardization, which could take some years to materialize. It also holds the potential to play a vital role in the CE marking process, offering Notified Bodies a comprehensive framework for evaluating medical device cybersecurity.
The introduction of AAMI SW96 marks a significant step forward in enhancing the security of medical devices, protecting patient safety, and upholding the integrity of healthcare technology. As the healthcare industry embraces technological advancements, cybersecurity standards like SW96 are crucial in ensuring that innovations benefit patients while minimizing risks. AAMI’s commitment to excellence in healthcare technology management shines through in this groundbreaking standard, setting a new benchmark for the industry.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #MedSysCon #FDA #cybersecurity
For further information please get in touch with us:
+49-176-57694801

22 May
3 Key Benefits Of Deploying Edge AI At The Point Of Care
3 Key Benefits Of Deploying Edge AI At The Point Of Care
Geoffrey Sheir, an edge inference expert at PA Consulting and Dan Talmage, a medical device development expert at PA Consulting wrote in the latest issue of meddeviceonline.com: “The point of care is shifting, with monitoring, diagnosis, and treatment taking place increasingly outside the conventional hospital setting and moving into patients’ homes. This is an exciting shift in the paradigm of healthcare.
The arrival of cloud computing has allowed industries to accelerate data processing by leveraging its immense scalability. Medtech companies have deployed artificial intelligence and machine learning algorithms in the cloud, where there is virtually unlimited processing power. This has improved patient outcomes, for example, through improved diagnostics and digital biomarkers. However, cloud computing’s reliance on the transfer of data to and from remote servers has some limitations, preventing many applications from shifting to the home setting.
The remoteness of the servers causes the first issue: latency. Real-time processing and turnaround time are often critical in rapid decision-making. Unpredictable load on the available resources can cause unpredictable timings on turnaround of processed data, potentially even changing the result of the processing.
The second issue is that cloud computing often requires a continuous connection to remote servers to exchange information. This reliance on an uninterrupted data pipe means any healthcare solution depending on cloud compute for AI algorithms will only work effectively in well-connected locations.
Last is data security; a patient’s medical information, according to laws such as HIPAA and GDPR, must adhere to strict handling and privacy requirements. This raises the concern of how to safely train and use algorithms running on third-party hardware and securely send data over networks. Such concerns have slowed the adoption of cloud-based AI in the medical field.
With a growing demand for AI in medtech applications, deployment of technology that can mitigate these risks must be considered for future applications. Edge inference is the practice of deploying machine learning (ML) models directly onto devices, allowing the data to be captured and processed at the point of care, rather than in the cloud. Processing data at the edge enables real-time processing, a reduced reliance on network quality, and increased data security, and the availability of specialist hardware means that edge inference is viable now. By identifying situations where these benefits can be obtained, medtech companies can use edge inference to shift the point of care.
Bring Care To The Front Line
Devices running edge inference computing enable real-time diagnostics to be performed outside specialist centers, reducing the time to treatment. The biggest opportunities exist for conditions where diagnoses are relatively accurate, but where patient outcomes are poor because diagnoses are not being done in a timely manner.
For example, a handheld retinal camera developed in Taiwan uses edge inference to diagnose diabetic eye disease, allowing primary caregivers to perform diagnoses that typically would be done by an ophthalmologist. The accuracy of the diagnostics provided is comparable to a specialist but is 10 times quicker than a competing cloud-based offering and does not require transport to a specialist ophthalmologist. Bringing these capabilities to the point of care not only reduces the risk and time taken for transport but also makes specialist care more accessible in primary care facilities without resident specialists.
Edge inference can also act as a real-time decision support tool for medical procedures. Virgo Surgical Video Solutions used edge inference in an endoscopy demo, detecting pre-cancerous growths with a latency of 17 ms, which is unachievable by beaming the data to and from the cloud. For GE HealthCare’s X-ray machines equipped with their Critical Care Suite ML algorithms, the X-ray machine automatically measures endotracheal tube positioning within seconds, allowing physicians to correct positioning errors in real time. This could feasibly extend to other procedures involving the insertion of medical devices into the body, such as stents, as well as more advanced procedures such as surgery.
Edge inference will ultimately enable closed-loop systems that perform monitoring, diagnostics, and treatment all in real time, reducing the need for manual intervention. Closed-loop intervention systems already exist for diabetes, like Medtronic’s MiniMed 780G insulin pump, where predictive algorithms monitor a patient’s insulin levels and perform interventions accordingly. We can extend this to a huge range of therapeutic areas and conditions that might require real-time interventions based on continuous monitoring, such as sepsis, cardiovascular activity, and seizure, where standard of care can be elevated across the board through the deployment of edge inference.
Reduce Reliance On High-Quality Networks
While solutions that process all the data in the cloud exist, maintaining data quality from the edge requires sufficient network bandwidth and reliability. This is by no means a guarantee, as the last mile of connectivity is governed by local internet service providers (ISPs). By processing data locally, edge inference reduces reliance on bandwidth, allowing care to be delivered in primary care facilities and in the home (e.g., on ambulatory monitoring systems) where network quality is not guaranteed.
Applications that capture data from body worn devices are a great example. Transmission of all data for cloud processing can take time, leading to a delayed diagnostic, reduced battery life, and increased data costs. Engineers should be considering whether early stages of the AI pipeline could run locally on a device, as this will significantly mitigate the challenges.
Additionally, opportunities to leverage edge inference can be found where care capabilities are not reaching as many people as they could be due to a lack of strong network infrastructure. For the handheld retinal camera, the fact that processing is done on-device allows for diagnoses to be made in facilities without an internet connection with reliably high bandwidth. The reduced reliance on network connectivity improves the scalability of solutions and allows smaller-scale primary care centers, or even visiting clinicians to the home, to deploy the same diagnostic capabilities as hospitals with a stronger network infrastructure.
Keep Patient Data Secure And Compliant
By processing data on the device and reducing the amount of data sent over the network, edge inference can help maintain data security for medical devices, so that compliance with data security regulations, such as HIPAA in the U.S. and GDPR for the EU, can be managed more easily compared to cloud processing. This can be leveraged particularly where there is a large stream of continuous data that needs to be kept secure, such as, for example, in monitoring devices such as wearables.
Video data can contain personally identifiable information and is a data type that is particularly important to safeguard. Care.ai created a HIPAA-compliant room-based patient monitoring device, which monitors patient behavior and alerts the clinicians when an adverse event is detected, such as, for example, if the patient falls or is at risk of developing pressure ulcers by sleeping in one position for too long. No video data is transmitted offboard for processing, preserving the patient’s privacy.
Other types of data, such as physiological data, may not be personally identifiable right now, but there is a similar need to keep this data secure to protect against compromise by future techniques. This is particularly pertinent to the raft of wearables performing continuous monitoring where a huge range of physiological data may be collected alongside personally identifiable data (e.g., GPS data). By reducing the amount of data these devices need to send over the network, edge inference helps keep this secure, too.
Edge Inference Is Viable Now
Edge inference is viable now, enabled by hardware and software advancements that make development and production easy. Chip vendors, such as NVIDIA and Coral, are providing a wealth of choices for hardware platforms on which to build edge inference solutions, such as dev kits to get development started quickly, as well as System on Modules with the versatility to build custom production systems. These platforms are also able to run on low power budgets, packing machine learning processing into small footprint, low-power devices.”
Please find the complete paper here.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #MedSysCon #FDA #AI
For further information please get in touch with us:
+49-176-57694801

20 Apr
The Evolving FDA Regulatory Landscape of Artificial Intelligence
The Evolving FDA Regulatory Landscape of Artificial Intelligence
Jesse Atkins, J.D., R.A.C., provided insights into this topic in the current issue of gardner.law: “
As artificial intelligence technology evolves on a seemingly minute-by-minute basis, the U.S. FDA’s regulatory approach continues to evolve in an effort to keep pace.
“The promise of artificial intelligence in medicine is to provide composite, panoramic views of individuals’ medical data; to improve decision making; to avoid errors such as misdiagnosis and unnecessary procedures; to help in the ordering and interpretation of appropriate tests; and to recommend treatment.” – Eric Topol, Deep Medicine: How Artificial Intelligence Can Make Healthcare Human Again
Whether it’s asking ChatGPT to rewrite your resume, getting behind the wheel of a self-driving vehicle, trusting the cybersecurity protocols of the cloud-storage platform to which you’re saving your family vacation photos, or teeing off on the first tee of your local country club,[1] if you’re reading this article, it is safe to assume that you interact with artificial intelligence and machine-learning (AI/ML) products on a daily basis.
Given the ubiquity with which AI/ML touches so many aspects of our lives, it should come as no surprise that the medical device industry has seen a rapid expansion of applications of AI/ML. Whether AI/ML functions as Software as a Medical Device (SaMD), or whether it is used in the development, clinical investigation, post-market data analysis, or quality control of medical products and their quality management systems, the proliferation of AI/ML in the healthcare industry is showing no signs of slowing down.
In an attempt to ensure adequate regulatory framework necessary to evaluate these products exists, the U.S. Federal Food and Drug Administration (FDA) has been active in developing action plans, engaging industry, issuing guidance documents, and devoting an increasing number of resources to the oversight of this growing discipline. This article summarizes FDA’s activities to date and highlights considerations medical device manufacturers should keep in mind as they develop AI/ML technologies.
[1] Yes, the driver in your golf bag was likely developed using artificial intelligence.
AI/ML in Medical Devices
Artificial intelligence is defined as “a branch of computer science, statistics, and engineering that uses algorithms or models to perform tasks and exhibit behaviors such as learning, making decisions, and making predictions.”[2] Machine learning is “a subset of AI that allows ML models to be developed by ML training algorithms through analysis of data, without models being explicitly programmed.”[3] AI/ML-enabled medical devices are those products intended to treat, cure, prevent, mitigate, or diagnose disease that use AI/ML, in whole or in part, to achieve their intended medical purpose. AI/ML-enabled devices have the potential to reduce the cost of medical intervention, to improve efficiency and early-detection capabilities of diagnostics, and to improve health outcomes and evaluation of patient progress through real-time analytics.
FDA, which regulates and grants marketing authorization for medical devices via one of three premarket pathways (premarket approval (PMA), De Novo Classification, or 510(k) clearance), approved its first AI/ML-enabled medical device in 1995 with the PMA approval of the PAPNET Testing System (P940029), a semi-automated test indicated to aid in the rescreening of cervical smears previously reported as negative and intended to detect cervical epithelial abnormalities, to be used to support clinician decision-making. Two decades later, FDA granted De Novo classification of the first autonomous AI/ML-powered diagnostic platform, the IDx-DR software algorithm, intended for early detection of diabetic retinopathy.
Today, FDA maintains a database of AI/ML-enabled medical devices, which it periodically updates, in order to showcase the growing number of medical devices authorized by the agency. As of the most recent update, 521 AI/ML-enabled medical devices have been granted marketing authorization by FDA, with the vast majority of devices falling into radiological and cardiovascular devices categories.
[2] Shawn Forrest, CDRH Digital Health Center of Excellence, Artificial Intelligence/ Machine Learning (AI/ML)-Enabled Medical Devices: Tailoring a Regulatory Framework to Encourage Responsible Innovation in AI/ML, https://www.fda.gov/media/160125/download (Adapted from IMDRF Artificial Intelligence Medical Devices Key Terms & Definitions Final document, posted May 9, 2022 at: https://www.imdrf.org/documents/machine-learning-enabled-medical-devices-key-terms-and-definitions).
[3] Id.
FDA’s Regulatory Approach to AI/ML
FDA is on-record as making AI/ML oversight and rulemaking a focus area, with the most significant efforts emanating from the Center for Devices and Radiological Health (CDRH) Digital Health Center of Excellence, originally launched in 2020.
FDA’s most significant foray into the AI/ML arena came in April 2019, when the agency issued the “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD),” a discussion paper and request for feedback issued to industry. In the discussion paper, FDA elicited stakeholder feedback on a number of topics, including AI/ML-enabled SaMD premarket review, regulatory evaluation of post-market modifications to AI/ML-enabled SaMD, and quality considerations, including Good Machine Learning Practices (GMLP) principles. FDA stressed the importance of consumer transparency and effective ongoing real-world performance monitoring for AI/ML-enabled devices.
In response to a significant volume of stakeholder feedback to the 2019 proposed framework, FDA published its “Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) Action Plan” in January 2021. In the action plan, FDA committed to a five pillars of its ongoing approach to regulating AI/ML-Based SaMD:
- Tailored regulatory framework for AI/ML-based SaMD. FDA recognized the need for, and committed to the promulgation of guidance to support, a new paradigm for regulatory oversight of AI/ML-enabled devices, most notably with regards to postmarket change control.
- GMLP development. FDA committed to continuing efforts, both domestically and in concert with the international regulatory community, toward the development of harmonized GMLP principles that will guide product development and maintenance.
- Patient-centered approach incorporating transparency to users. Leaning on learnings from the agency’s Patient Engagement Advisory Committee meeting on AI/ML-enabled devices, FDA stressed the need for and committed to creating forums for the dissemination of information about AI/ML-enabled devices to the general public. Information relevant to FDA and patient-engagement stakeholders includes information about the source of data used in algorithm training and information to ensure the benefits, risks, and limitations of AI/ML-enabled devices are easily understood by all users.
- Regulatory science methods related to algorithm bias and robustness. FDA acknowledged that, while not unique to AI/ML-enabled devices, there are unique concerns related to bias and generalizability of use associated with these devices and the data forming the basis for their functions. FDA committed to supporting regulatory science research methods to ensure factors such as race, ethnicity, and socio-economic status are taken into account in the development and maintenance of AI/ML-enabled devices.
- Incorporation of real-world performance metrics. As real-world data continues to be an important topic in medical device development and regulation, FDA acknowledges that importance is only heightened based on the continuous learning and real-world-data-based algorithms of AI/ML-based devices. FDA committed to piloting real-world performance monitoring on a voluntary basis in order to develop uniform real-world performance principles applicable to AI/ML-enabled device oversight.
Since announcing its 2021 action plan, FDA has issued a number of guidance documents aimed at fulfilling the commitments related to the regulatory oversight of AI/ML-enabled devices contained therein. Key guidance documents are discussed below.
- Draft Guidance: Assessing the Credibility of Computational Modeling and Simulation in Medical Device Submissions (December 2021)
This draft guidance addresses the use of computational modeling and simulation in medical device submissions, proposing a framework by which this type of information can be gathered and presented in a manner that ensures reliability and credibility sufficient to either: (1) support regulatory approval or clearance; or (2) support the use of computational modeling and simulation within the medical device software itself. The draft guidance provides a generalized framework for assessing credibility of computational modeling in a hypothetical nine-step process. The principles of this guidance can be applicable both to the use of AI/ML in computational modeling and simulation activities associated with product development, as well as in supporting the premarket approval or clearance of computational modeling and simulation functions of AI/ML-enabled devices.
- Draft Guidance: Computer Software Assurance for Production and Quality System Software (September 2022)
This draft guidance proposes a risk-based approach to computer software assurance activities related to features, functions, and operations of production and quality system software that present a high process risk (and, accordingly, medical device risk). The draft guidance provides insights into considerations manufacturers should account for in qualification and validation of quality system and manufacturing software, as well as in the documentation rigor that should be employed in these activities. As AI/ML-enabled product development continues to become more and more mainstream, these risk-based approaches offer insight into satisfactory means of testing and documenting these systems.
- Final Guidance: Clinical Decision Support Software (September 2022)
The final Clinical Decision Support (CDS) Software guidance was received by industry as a significant departure from the original draft guidance of the same name (originally issued in September 2019). Centrally, the final guidance provides the rubric by which FDA will determine whether a software function is considered a CDS function (exempt from FDA oversight) or a medical device function (requiring the manufacturer to comply with FDA regulations and premarket notification or approval requirements). The approach described in the final guidance is comprised of four criteria (where a software function is excluded from the statutory definition of a medical device only by meeting all four criteria): (i) the software function is not intended to acquire, process, or analyze a medical image or signal from an in vitro diagnostic or a pattern or signal from a signal acquisition system; (ii) the software function is intended for the purpose of displaying, analyzing, or printing medical information; (iii) the software function is intended for the purpose of supporting or providing recommendations to an HCP about prevention, diagnosis, or treatment; and (iv) the software function is intended for the purpose of enabling a healthcare practitioner to independently review the basis for the recommendations (in a manner that the practitioner does not rely primarily on any software recommendations to make a diagnosis or treatment decision). Developers of software functions, particularly those powered by AI/ML who previously assessed their products to be exempt from FDA oversight, should carefully evaluate each software function to ensure their product does not inadvertently assume an intended use classifying it as a medical device.
The landscape of FDA oversight of AI/ML-enabled medical devices is dynamic, and the only constant that industry should expect in the coming years is change. Evidence of this change can be found in the recently announced CDRH Proposed Guidances for Fiscal Year 2023. Draft guidance topics for the coming fiscal year most notably includes “Marketing Submission Recommendations for A Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions.” Additional guidances related to cybersecurity, content of premarket submissions for device software functions, and evaluation of sex-specific and gender-specific data in medical device clinical studies are sure to impact manufacturer activities related to the development, regulatory submission, and maintenance of AI/ML-enabled medical devices.”
Please find the complete article here.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #MedSysCon #FDA #AI #Artificial_Intelligence
For further information please get in touch with us:
+49-176-57694801

14 Mar
The Buzz About Medical Device Cybersecurity in 2022 and Beyond
Paul Rothermel, Associate Attorney, Gardner Law, shares insights in https://gardner.law/alerts: “Medical device cybersecurity continues to create buzz, as the FBI issues a Private Industry Notification to health care providers outlining cybersecurity risks for medical devices. This follows FDA’s released Draft Guidance and FDA recommendations for questions health care providers should ask manufacturers about medical device cybersecurity. Click here to read a prior alert on this guidance.
Medical device cybersecurity is critical to patient safety and continues to attract attention from regulators, industry and even law enforcement. Unlike the imagined threats faced by our favorite space-bound toy with a badge, the threat of medical device compromise is very real and federal law enforcement has taken note.
A September 2022 Private Industry Notification issued to health care providers from the FBI identified some key risk areas:
- Default configurations and passwords;
- Devices with delayed vulnerability patching;
- Devices designed without security in mind;
- Internet of Things/connected devices;
- Specific device types, such as insulin pumps, intracardiac defibrillators, mobile cardiac telemetry, pacemakers, intrathecal pain pumps; and
- End of life/legacy devices with minimal security patching/upgrades
This closely follows the recent FDA Draft Guidance which also emphasizes the impact of device connectivity to safety and effectiveness of medical devices. This guidance released April 8, 2022 is intended to replace 2018 draft guidance and includes investigational devices in its scope with the comment period closing on July 7, 2022. It emphasizes risk management throughout the “Total Product Life Cycle” to address device cybersecurity, including:
- Designing medical devices for cybersecurity (implementing cybersecurity as part of “software validation and risk analysis” as appropriate);
- Monitoring, identifying, and addressing cybersecurity vulnerabilities in devices that are on the market; and
- Outlining recommended content for premarket submission re: cybersecurity and encourages use of a Secure Product Development Framework.
FDA states in the Draft Guidance:
Software validation and risk analyses are key elements of cybersecurity analyses and demonstrating whether a connected device has a reasonable assurance of safety and effectiveness. FDA requires manufacturers to implement development processes that account for and address cybersecurity risks as part of design controls (21 CFR 820.30). For example, these processes should address the identification of security risks, the design requirements for how the risks will be controlled, and the evidence that the controls function as designed and are effective in their environment of use for ensuring adequate security.
How can manufacturers address device cybersecurity
What follows are some suggestions based on FDA draft guidance. While the guidance is still in draft, thus not binding, it signals FDA’s current thinking with regard to medical device cybersecurity and will help manufacturers better prepare to engage with FDA and with their customers on the topic of cybersecurity.
Design your device for cybersecurity
Companies should address security objectives such as maintaining authenticity, authorization, availability, confidentiality, and secure and timely updates/patching for the devices. FDA notes that “exploitation of known vulnerabilities or weak cybersecurity controls” is reasonably foreseeable and must be addressed in the device design. FDA also warns manufacturers that inadequate cybersecurity controls “may cause a device to be misbranded […] among other possible violations…”
Address cybersecurity risk in premarket submissions
Address key security objectives and then describe how the device design actually addresses and integrates these security objectives based on the particular device (e.g., intended use and indication, electronic data interfaces, intended/actual environment of use, types of cybersecurity vulnerabilities present, exploitability of vulnerabilities, and risk of patient harm from exploited vulnerabilities.
Offer transparent cybersecurity information to device users
FDA suggests information should be provided about integrating device into the use environment, maintaining device cybersecurity over its lifecycle, and any information potentially affecting safety and effectiveness of the device. FDA also proposes that interconnected devices should include cybersecurity information in device labeling. The draft guidance suggests providing insufficient cybersecurity information to device users may compromise device safety and effectiveness.
Security risk management
FDA recommends separating safety and security risk assessments, noting that they focus on different things: safety risk assessment emphasizes probability of harm while security risk assessment focuses on exploitability and to “expose how threats, through vulnerabilities, can manifest patient harm and other potential risks.” Identified risks should be mitigated comprehensively in the design (or if not possible, in compensating controls). In cases where risks are only partially or not at all mitigated, they should be assessed further. As a last resort, risk transfer to users or even the patient may be necessary but these risks should be known, assessed, and communicated appropriately. Continue to identify, assess, and mitigate cybersecurity vulnerabilities throughout the entire lifecycle of the device using the company’s “Secure Product Development Framework”. Document, document, document – summarize risk evaluation methods, processes, details of assessments and mitigation undertaken. The risk management process must be traceable.
Other considerations
Customer awareness is increasing
FDA recently posted a video for “Cybersecurity Awareness Month” that recommends health care providers ask device manufacturers questions about cybersecurity, including:
- How is the device updated?
- What does it connect to?
- What happens if the connection is unavailable?
- What are the cybersecurity risks associated with the device?
- What cybersecurity resources do they have to support your patients?
- Who should you reach out to with questions if you have a concern?
Provider’s asking about device cybersecurity is not a new trend. Health care providers already have been increasing scrutiny of devices in recent years. But it does point to a future where medical device cybersecurity questionnaires are normative even for smaller providers.
Privacy remains a concern
If your medical device interacts with patient information, the Health Insurance Portability and Accountability Act (“HIPAA”), Federal Trade Commission (“FTC”) Act, or other laws may apply. This is especially critical for devices that have connectivity to systems managed by the device manufacturer. By designing your medical device for privacy and data security upfront and understanding the implications of your design choices when the device goes to market or enters a clinical trial phase, you will be better prepared for compliance with these requirements.”
Please find the complete article here.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #MedSysCon #cybersecurity #FBI
For further information please get in touch with us:
+49-176-57694801

17 Feb
Top ten observations from 2022 in life sciences digital and analytics
Top ten observations from 2022 in life sciences digital and analytics
Bjorn Albrecht a partner in McKinsey’s Paris office; Siméone de Fremond a consultant in the Geneva office, Thomas Devenyns an associate partner; Richard Ting Li a consultant in the New York office; Dan Tinkoff a senior partner in the New Jersey office; and Lieven Van der Veken a senior partner in the Lyon office wrote an enlightening article at mckinsey.com:
“Life sciences companies are making progress in adopting and deploying digital and analytics. But they can go further and faster by heeding some lessons from the past few years.
Looking toward the new year is a good time to take stock of the life sciences industry’s digital maturity. This article provides ten observations from McKinsey’s work in this space, complemented by targeted analyses and a McKinsey survey conducted in November 2022 of one hundred digital and analytics (DnA) leaders in life sciences functional areas, such as R&D, manufacturing and supply chain (M&SC), commercial, and enabling functions such as finance and HR.4 Life sciences companies have historically lagged behind banking, telecom, retail, and other industries in digital maturity. A McKinsey analysis from 2016 identified a multitude of factors that make life sciences companies slow to adopt DnA innovations, including not fully understanding healthcare practitioners’ (HCPs) decision journeys, challenges linking DnA to the broader business, and difficulty maintaining an efficient operating model. Has the pandemic accelerated the adoption of digital technologies, have life sciences companies caught up at all, and how far are they from realizing the full potential of digital and analytics?
The results are sobering. The gaps with other industries are not closing, and the reported benefit to businesses is moderate. All players in the ecosystem can benefit from incorporating digital and analytics into every aspect of their operations to fully deliver on the promise for patients and societies. Here are our ten observations in detail.
Where the industry is now
1. The digital maturity gap persists between life sciences and digital leaders. Despite efforts made over the past few years, life sciences companies still trail cross-industry leaders in digital maturity by a factor of two to three times in every key dimension—strategy, culture, organization, and capabilities—without any clear signs of catching up.
Pharmaceutical companies (pharmacos) surveyed are slightly more mature than medical technology (medtech) companies: 55 percent of pharma DnA leaders say they have deployed some applications at scale, compared with 34 percent of medtech DnA leaders. Overall, commercial and R&D are more digitally mature than other functions, with more than 14 percent of DnA leaders in those areas reporting that digital applications are deployed at scale and are contributing tangible business value. Less than 6 percent of DnA leaders see the same in M&SC and enabling functions.
Life sciences has also struggled to execute on the idea of building novel digital businesses beyond digital solutions like apps supporting their existing products. The jury is still out on whether life sciences companies are the best owners of these, due to misalignment of incentives, the need for agility, and the capabilities needed.
2. Life sciences companies are starting to capture value from digital and analytics but are still just scratching the surface. Life sciences DnA leaders surveyed estimate that digital and analytics drove a 5 to 15 percent bottom-line improvement in specific pockets of their functional areas over the past five years, yielding an annual global impact of $6 billion to $9 billion. This compares with an estimated $130 billion to $190 billion that the full application of digital solutions and innovation along the life sciences value chain could bring.
The largest opportunities DnA could help unlock are derisking drug discovery, accelerating clinical trials, and reinventing engagement with HCPs. As a result, many leading companies are incorporating DnA into early-stage drug discovery and clinical development to shrink timelines and improve the probability of success. In addition, leading players are reinventing their interactions with healthcare providers and patients to provide enhanced and tailored experiences and achieve better treatment outcomes.
3. Early innovation is clearly catching up, but it took the top 40 life sciences companies until 2021 to overtake the big three tech companies (Alphabet, Amazon, and Apple). The top 20 pharmacos and the top 20 medtech companies generated more than 1,700 DnA and life sciences–related patents in 2021, a growth surge of more than 70 percent over 2017 that finally allowed the industry to overtake the combined volumes of Alphabet, Amazon, and Apple. Publications also increased by 28 percent in that period, reaching 650 last year.
In other industries, such as retail banking, strategic and financial benefits accrue to digital innovators. Still, despite the recent burst in digital innovation, there is not yet a clear correlation between life sciences companies’ digital maturity and their financial performance. This is probably because of long product development timelines and limited short-term benefits, especially in R&D, underlining the importance of defining and linking digital strategy to scientific and business value at every level of the organization.
4. US venture funding in digital health peaked during the COVID-19 pandemic and is now stabilizing at twice its historical base, with the big three tech companies’ investments edging out those of the top 40 life sciences companies. Funding along the life sciences value chain jumped from $5 billion in 2019 to more than $20 billion in 2021, with more than 45 percent of the funding in commercial solutions. Funding in research and development ranked second, totaling $6.4 billion, with almost equal interest from investors in research and early development ($3 billion) and development, regulatory, and safety ($3.4 billion).
Year-to-date funding and digital-health stock indexes, such as the MSCI ACWI IMI Digital Health Index, for 2022 have almost halved compared with 2021, partially because of a general public-market correction. However, they are still twice their 2019 levels.
The US life sciences sector still leads Europe and Asia in DnA investments. The top 20 pharmacos and the top 20 medtech companies made slightly more DnA-related major deals in the United States than in the European Union over the past five years—32 versus 26— but on average, the US deals were about five times larger, $700 million versus $150 million in the European Union. This is a strong indicator of the difference across continents in the level and pace of innovation.
Globally, the big three tech players are edging out the top 40 life sciences companies as leading strategic investors. The top 40 life sciences players have invested almost $20 billion in DnA deals since 2017, but less than $6 billion in the past three years; big tech players invested $10 billion in tech-related healthcare deals, $8 billion of that in the past three years. Big tech players can leverage their ability to generate cash flow to fuel their M&A activities, while life sciences companies tend to prioritize asset acquisitions over DnA deals.
Where the industry is going
5. Hiring and spending are accelerating, but not by much. The hiring rate of DnA talent by life sciences companies is accelerating slightly, from 2 to 4 percent a year over the past five years to an expected 3 to 5 percent a year over the next three. Only 10 percent of respondents said they had grown the talent in their functional areas by more than 30 percent, and only 9 percent said that they are targeting such an expansion over the next three years. Hiring is generally more robust in R&D and commercial, and it’s expected to remain so.
Similarly, leaders expect to grow their DnA spending by another 10 to 15 percent over the next three years, starting from today’s baseline of 8 to 12 percent of their function’s budgets. Only 7 percent of leaders expect to increase their investment by more than 30 percent over the next three years.
6. Analytics rule the world of life sciences tech investments. Life sciences companies make 45 percent of their tech investments in three analytics-related technologies—applied artificial intelligence (AI), industrialized machine learning (ML), and cloud and edge computing—and expect to derive most of their short- to medium-term benefits from them.
Applied AI has permeated the life sciences value chain, allowing more business decisions to be made based on data the organization has generated or acquired. Cloud and industrialized ML allow life sciences companies to adopt new AI approaches more reliably and at a faster pace and larger scale, accelerating tech-driven innovation.
Cloud technologies are increasingly being embraced by life sciences to speed tech-enabled business transformations; in 2021, more than 80 percent of the top 20 global pharma and medtech companies were operating in the cloud. In addition, companies are increasingly leveraging the cloud to shift IT infrastructure management and to fill gaps in their software development and analytics capabilities, buying software as a service rather than building and operating it themselves.
Despite the high media attention, longer-term bets such as quantum technologies, Web3, advanced connectivity, and immersive reality together account for only about 15 percent of life sciences tech investments.
Beyond focusing on a single tech trend, the research shows that DnA leaders could consider investing in a combination of trends to unlock the most innovation and new sources of value. For instance, life sciences companies could accelerate drug discovery, create more personalized treatments, and optimize treatment plans for patients by leveraging a combination of Al and ML, cloud computing, quantum technologies, and bioengineering.
7. The plan for the future is largely the plan of today. DnA leaders are betting that the areas that have brought the most value to date will be the primary value drivers for the next few years.
In the past three to five years, each function has had two or three areas in which digital and analytics brought noticeable changes and moderate impact. These included disease state and target understanding in research and early development, clinical trial planning and execution in development, regulatory and safety, end-to-end supply chain in M&SC, go-to-market and field force effectiveness in commercial, unmet medical need in medical, and shared services productivity in enabling functions.
What it will take to succeed and scale
8. The challenge has shifted from strategic alignment and executive leadership to data and talent. The top hurdle to scaling life sciences DnA in 2020 was a lack of leadership support and misaligned strategy, but that is no longer the case. Instead, the key challenges today include the following:
- Lack of high-quality data sources and data integration. This challenge is cited by both pharma and medtech DnA leaders as a top issue. In addition to accessing existing data, DnA leaders are increasingly interested in supporting decision making by generating new data from business operations, such as new digital channels for HCP engagement.
- Lack of the right DnA talent. To scale DnA transformations successfully, life sciences companies need cross-functional teams with experienced leaders and accomplished DnA practitioners. In addition, life sciences companies still require the role of translator, because most DnA experts don’t have degrees in life sciences or prior knowledge of a specific therapeutic area. The reverse is true for colleagues on the business side. It is, therefore, critical to have experts who can bridge the gap between the business and analytics teams to facilitate cross-functional collaboration.
- Lack of adoption and scaling. In many functional areas, DnA solutions still are not deployed at scale. In R&D, for example, many companies are still producing proofs of concepts and pilots detached from business needs without a plan or the means to scale them. In addition, many life sciences companies fail to recognize the need to transform the mindset and culture of their organizations to drive DnA adoption at scale.”
Please find the complete article here.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #MedSysCon #2022
For further information please get in touch with us:
+49-176-57694801
Topics:
#healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #medsyscon

05 Feb
The health benefits and business potential of digital therapeutics
The health benefits and business potential of digital therapeutics
Chirag Adatia a partner in McKinsey’s Gurugram office, Samarth Shah consultant. Ralf Dreischmeier senior partner in the London office and Kirtika Sharma, partner in the Mumbai office wrote an enlightening article at mckinsey.com:
“Digital-native start-ups and healthcare incumbents can both play important roles in building and scaling digital therapeutics to improve the management of chronic health conditions.
Around the world, the burden of chronic disease is increasing at a rapid pace. Unfortunately, most of these conditions are irreversible and need to be managed through lifelong medication use. However, many patients struggle with adhering to prescribed medications and implementing the behavioral and lifestyle changes that are needed to manage their diseases and stabilize their conditions. Often, physicians and other healthcare providers have little ability to monitor the extent to which patients are following their recommendations and maintaining treatment regimens. As a result, disease burdens at a population level are higher than they should be.
These challenges have created a need for comprehensive disease management solutions that are best enabled by digital technologies. In 2021, global digital health funding grew 79 percent over the previous year to reach $57.2 billion. Much attention and funding have flowed toward digital therapeutics, which can include multiple points of intervention along the patient journey, including monitoring, medication adherence, behavioral engagement, personalized coaching, and real-time custom health recommendations. Within digital health, funding for digital therapeutics (including solutions for mental health) has grown at an even faster pace—up 134 percent from the prior year to reach $8.9 billion in 2021.
The impact potential here is significant, both in terms of clinical outcomes and economic benefits for stakeholders and societies. For example, research has shown that digital disease management can drive a 45 percent reduction in the three-month rate of major adverse cardiovascular events (MACEs) and a 50 percent reduction in the 30-day readmission rates for patients after acute myocardial infarction (AMI). Similarly, it can help lower hemoglobin A1c (HbA1c) levels by one percentage point among patients with type 2 diabetes. These data points illustrate the extent to which digital disease management can help save lives while also keeping patients healthier, which reduces costs for many stakeholders, including the patients themselves.
Research has shown that digital disease management can drive a 45 percent reduction in the three-month rate of major adverse cardiovascular events (MACEs) and a 50 percent reduction in the 30-day readmission rates for patients.
Many players are trying to disrupt the disease management space and develop new innovative models to manage chronic diseases. New-age start-ups bring radical, unconstrained perspectives, while incumbents contribute a much more detailed understanding of the challenges and various stakeholders. Ultimately, both start-ups and incumbents have critical roles to play in disrupting the space and scaling up solutions.
Digital therapeutics can play an important role in chronic-disease management
The burden of chronic diseases has been increasing globally and is expected to continue. Chronic diseases (such as cardiovascular disease, cancer, diabetes, and respiratory disease) were causes or contributing factors in 75 percent of worldwide deaths in 2010 and 79 percent in 2020. By 2030, experts predict that chronic diseases will contribute to as much as 84 percent of total global mortality.
Poor monitoring of and adherence to prescribed medications undermine the management of chronic diseases. According to a 2021 global study, compliance among patients with type 2 diabetes ranges from 69 to 79 percent.
Of course, chronic diseases need to be managed not only by medication but also with regular monitoring and lifestyle changes. Hence, providers need better end-to-end solutions that proactively and comprehensively monitor patient health, as well as encourage behavioral changes to improve adherence to prescribed medications, diet, and lifestyles.
Digital technologies can play an important role in improving disease management by tackling these challenges. The potential for digital therapeutics to have a big impact is evidenced by the fact that almost two-thirds of the global population now has internet access.
Research has shown that digital solutions for disease management can drive better outcomes for patients living with chronic diseases. Examples include the following:
- A study of ten thousand patients by the Poland National Health Fund showed a 45 percent reduction in three-month MACE rate and a 40 percent reduction in 12-month mortality rate achieved through managed care after AMI. The study involved cardiac rehabilitation with physician guidance, counseling sessions on lifestyle modification, education on the associated risk factors, therapy, and in-person relaxation sessions.
- A study by the Mayo Clinic in partnership with Healarium showed a reduction in three-month rehospitalizations and emergency department visits of 40 percent for patients following AMI, a weight reduction of 4.0 kilograms, and a 10.8-millimeter reduction in systolic blood pressure. The study involved tracking of vitals, diet, and physical activity, setting reminders and goals, information on current health status, and educational courses for patients.
- A US study of more than one thousand patients by Johns Hopkins and Corrie Health showed a 50 percent reduction in the 30-day readmission rate in patients following AMI attained through digital-health-based interventions. The study involved continuous monitoring of vitals with the help of connected devices; educational content on procedures, risk factors, and lifestyle modifications; medication management through reminders and tracking adherence; connection with the care team; mood tracking; and the ability to check the side effects of medication.
- A one percentage-point reduction in HbA1c levels was shown in patients with type 2 diabetes who participated in an online patient community as part of Virta Health’s ten-week nonrandomized parallel arm study with 262 outpatients. The patients were given individualized nutritional recommendations through dedicated health coaches, continuous glucose monitoring kits, and online counseling with doctors.
Eight key elements of impactful digital therapeutics solutions
Strong digital therapeutics solutions typically contain most or all of the following eight elements:
- Regular monitoring, measurement, and feedback through connected medical devices. Devices such as smart inhalers for respiratory conditions or continuous glucose monitors for diabetes can provide patients with nudges and alerts for out-of-range readings. For example, Boston-based Biofourmis applies digital therapeutics through the continuous monitoring of connected medical devices. The company offers a doctor-prescribed digital platform approved by the US Food and Drug Administration for patients suffering from chronic heart conditions. Its unique wearable devices offer specialty chronic heart care management, including automated medication management combined with a multidisciplinary remote clinical-care team. In 2022, the company was valued at $1.3 billion.
- Keeping payers and providers in the loop. When patients grant access to their vital statistics, insurance companies, caregivers, and employers can reward them for progress in stabilizing or improving chronic health conditions. For example, Livongo, a program from Teladoc Health, allows patients with diabetes to monitor their condition regularly and send alerts via Bluetooth to an app on their own and their caregiver’s phones if readings exceed normal ranges. Over time, patients enrolled with Livongo have achieved a 0.8 percentage-point drop in HbA1c for diabetics, a 10.0-millimeter hemoglobin drop in blood pressure for patients with hypertension, a 1.8-point drop in body mass index, and a 7.0 percent drop in weight. Livongo allows payers and providers to identify and reward good behavior, as well as deter or penalize poor adherence to health plans prescribed by providers.
- Personalized coaching and support. Patients can connect with specific coaches to obtain a personalized diet and exercise plan tailored to their chronic illnesses. This can be very effective from a therapeutic standpoint. A meta-analysis of digital health interventions on blood pressure management showed that digital counseling alongside antihypertensive medical therapy reduced systolic blood pressure by 50 percent relative to controls. For example, Hinge Health has built a $6.2 billion business that offers wearable sensors combined with personalized exercise therapy and one-on-one health coaching.
- Gamified behavioral modification. Digital therapeutics solutions can include gamified challenges and incentives to track and drive adherence to prescribed diets, lifestyle practices, and medications. For example, Discovery, a South African health insurance company, encourages its members to make healthier choices through its Vitality behavioral change program that combines data analytics with rewards and incentives for healthier lifestyle choices.
- Building a thriving community. An active virtual patient community can drive adherence by challenging and motivating patients to live up to their own health goals. For instance, one study of seven thousand patients with amyotrophic lateral sclerosis (ALS), multiple sclerosis, Parkinson’s disease, HIV, fibromyalgia, or mood disorders found that nearly 60 percent thought the PatientsLikeMe health network helped give them a better understanding of the side effects of medications. The study also found that nearly a quarter of patients with mood disorders needed less inpatient care thanks to their use of the PatientsLikeMe site.
- Health mall. A recent McKinsey survey found that 90 percent of healthcare leaders believe that patients interacting with digital health ecosystems want an integrated journey rather than point experiences or solutions. Healthcare companies can meet this desire for integration by offering digital health malls that include access to prescribed medications, health supplements, wellness products, and diagnostic tests at the click of a button.
- Patient education. Digital education materials can give patients and their family members information on disease conditions, treatment options, diet, and healthy lifestyle choices. For instance, the Midday app launched by Mayo Clinic and digital health start-up Lisa Health provides support, including educational content, to women experiencing menopause.
- Advanced analytics to predict and prevent health events. Organizations are working now to build data algorithms that could identify and predict triggers for healthcare events. They could suggest when to take preventative action or where lifestyle and behavioral changes might forestall adverse events.”
Please find the complete article here.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #MedSysCon #digitaltherapeutics #DTx
For further information please get in touch with us:
+49-176-57694801

15 Jan
The FDA & EU IVDR Regulatory Frameworks For IVD SaMD
The FDA & EU IVDR Regulatory Frameworks For IVD SaMD
Marcelo Trevino, global vice president, regulatory affairs and quality assurance at Agendia, a molecular diagnostics company focused on breast cancer genomic testing wrote in the current issue of meddeviceonline: “Software that is qualified as an in vitro diagnostic medical device (known as SaMD IVD) is affected by the same requirements as other medical devices, which are governed by various FDA regulations and EU In Vitro Diagnostic (IVD) Regulation EU 2017/746. Therefore, SaMD IVD’s intended use, classification, clinical/performance requirements, and the content of the technical documentation must comply with these regulations. In my last article, I described how to know if FDA and EU regulators qualify your software as an IVD SaMD. This article builds on that foundation and summarizes the regulatory framework required for IVD SaMD in the U.S. and European markets.
Defining Software’s Intended Use And Regulatory Classification In The U.S.
In the U.S., IVD SaMD is classified according to risk-based criteria based on the intended use, then software is categorized by level of concern to establish the evidence required. There are four steps involved in the classification:
1. Select the appropriate device type regulation: There are three sections of regulations that cover IVD devices: 21 CFR Part 862 Clinical Chemistry and Clinical Toxicology, 21 CFR Part 864 Hematology and Pathology, and 21 CFR Part 866 Immunology and Microbiology.
2. Determine the appropriate device risk classification: There are three levels: Class I (low to moderate risk, general controls required), Class II (moderate to high risk, general controls and special controls required), or Class III (highest risk, general controls and premarket approval required).
3. Review device classification exceptions: Depending on the applicable regulations associated with the device, some devices might be exempt from premarket notification requirements.
4. Determine the associated product codes: Based on the applicable regulation number, FDA’s product classification database shall be used to review available product codes to identify predicate devices and the substantial equivalence information.
The “level of concern” refers to an estimate of the severity of injury that a device could permit or inflict, either directly or indirectly, on a patient or operator as a result of device failures, design flaws, or simply by virtue of employing the device for its intended use. There are three levels of concern for device software in the U.S.: major, moderate, and minor. To establish the level of concern, manufacturers should answer questions provided in FDA’s Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices and establish a rationale. This will determine the amount of documentation required in the premarket submission. FDA considers IVD software to be at least a moderate level of concern because software flaws could indirectly affect a patient and could potentially result in injury.
Determining Software’s Intended Use And Regulatory Classification In The EU
After the software is qualified as an IVD, the manufacturer shall define its intended use, since the intended use determines the regulatory classification of the software. The intended use shall capture the software’s core functionality and the effect of the information provided for healthcare decisions.
Once the software is qualified as an in vitro diagnostic medical device, the next step is to determine its risk class based on Annex VIII of Regulation (EU) 2017/746.
Rule 1.4 of Annex VIII indicates: “Software, which drives a device or influences the use of a device, shall fall within the same class as the device; If the software is independent of any other device, it shall be classified in its own right.”
Rule 1.8 of Annex VIII indicates: “Where a manufacturer states multiple intended purposes for a device, and as a result the device falls into more than one class, it shall be classified in the higher class.” Rule 1.9. of Annex VIII indicates: “If several classification rules apply to the same device, the rule resulting in the higher classification shall apply.”
MDCG 2019-11 provides examples of classification. However, it is important to note that these examples should not be considered confirmation of the final device’s classification, as manufacturers shall consider all the rules under Annex VIII for the in vitro diagnostic medical device software according to its intended purpose, and the justification should be documented in the device’s technical documentation.
General Safety And Performance Requirements (GSPR) For In Vitro Diagnostic Medical Device Software In The EU
In the EU, once a software qualifies as an in vitro diagnostic medical device, the manufacturer must demonstrate compliance with the 20 general safety and performance requirements detailed in Annex I of Regulation (EU) 2017/746. If some of these are not applicable, a justification must be provided. Software compliance can be supported through the use of different harmonized standards such as EN ISO 14971:2019 for medical device risk management.
Below are the relevant GSPRs affecting software:
GSPR 13.2(d) requires devices to be designed and manufactured in such a way as to remove or reduce as far as possible the risks associated with the possible negative interaction between software and the IT environment within which it operates and interacts.
GSPR 16 focuses on software and includes four points:
- Software design must ensure repeatability, reliability, and performance in line with its intended use;
- Software must be developed and manufactured in accordance with the state of the art, taking into account the principles of development life cycle, risk management, including information security, verification, and validation;
- Software running on mobile platforms shall be designed and manufactured taking into account the specific features of the mobile platform (e.g., size and contrast ratio of the screen) and the external factors related to their use (varying environment as regards level of light or noise); and
- Manufacturers shall set out minimum requirements concerning hardware, IT networks characteristics, and IT security measures, including protection against unauthorized access necessary to run the software as intended.
GSPR 20.4.1 (ah) addresses information required in the instructions for use and requires that the instructions for use for devices that incorporate electronic programmable systems, including software, or software that are devices in themselves, shall contain minimum requirements concerning hardware, IT network characteristics, and IT security measures, including protection against unauthorized access, necessary to run the software as intended.
Software performance must also be demonstrated. MDCG 2020-1: Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software provides guidance on the level of evidence required according to the EU 2017/746 IVD Regulation.
Clinical Evidence Through Performance Evaluation: FDA And EU Requirements
FDA and EU IVDR regulators require clinical evidence through performance evaluation. FDA and EU recognize IMDRF/SaMD/WG/N41 Final: 2017 for demonstrating clinical evaluation of SaMD in general. In the EU, there is an additional guidance provided for IVD SaMD: MDCG-2020-1 Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software.
To demonstrate that the IVD SaMD performs as intended, manufacturers shall demonstrate that the software is designed using recognized standards and also that it can achieve its medical purpose safely and effectively. If the IVD SaMD uses an algorithm that must be trained to perform as intended, manufacturers shall provide a conclusion that the algorithm works for its medical purpose as part of the analytical and clinical performance evidence. Regulatory agencies in the U.S. and the EU will expect to see a description of the data sets used to evaluate the software, including inclusion or exclusion criteria, clinical site descriptions, number of subjects, algorithm development processes, statistical models used, and performance measures for verification and validation. Any claimed benefits must be supported with clinical evidence based on performance evaluation.
A valid clinical association shall be established between the IVD SaMD output and the target clinical condition and clinical studies to evaluate analytical and clinical performance. Effective performance of the SaMD’s intended use shall be demonstrated through analytical performance (demonstration that the software processes input data to generate accurate output data, which may include analytical sensitivity, analytical specificity, bias, precision, accuracy, limits of detection, and method comparison) and clinical performance (the analytical use of the software applied to the target population in a clinical care setting achieving its stated intended purpose or claimed benefit, which can be demonstrated referencing existing data from studies performed for the same intended use or by generating new clinical data for the specific software’s intended use).
EU IVDR And FDA Technical Documentation Requirements For Regulatory Review
For the EU, Annexes II and III of 2017/746 IVD Regulation provide details on the technical documentation that must be gathered for the software. EN 62304 provides good guidance to ensure most requirements are adequately addressed and the documentation generated according to EN 62304 standard allows a large part of the regulatory requirements to be met. The information to be provided includes, among others: an overview of the entire system and a description of the data interpretation methodology, a summary of the results of all verification, validation, and testing applicable in a real-world usage environment, labeling information, configurations, and operating systems required.
Similarly, FDA’s draft guidance Content of Premarket Submissions for Device Software Functions and Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices provide details on the recommended documentation that software manufacturers should include in premarket submissions. This includes, for example, software description, system and software architecture diagrams, risk management file, device hazard analysis for software devices, software design specifications, software level of concern classification, and verification and validation activities, among others.
FDA’s MDUFA Digital Health Commitments
As part of the Medical Device User Fee Amendment V (MDUFA V) passed by the U.S. Congress on Sept. 30, 2022, FDA established several Digital Health Commitments to be accomplished between fiscal year 2023 and fiscal year 2027 that will continue to build its digital health expertise and work to streamline and align their review processes with software life cycles for digital health products. The following are the six Digital Health Commitments:
- Continue to develop software and digital health technical expertise to provide assistance for premarket submissions that include digital health.
- Strengthen efforts to expand staff understanding of digital health topics and enhance consistent evaluation submissions.
- Continue to participate in international harmonization efforts related to digital health.
- Finalize the draft guidance Content of Pre-market submissions for device software functions by 18 months from close of the comment period.
- Publish draft guidance describing a process to evaluate a predetermined change control plan for digital health devices.
- Engage with stakeholders including patients, users, and industry through roundtables, informal meetings, and teleconferences to explore regulatory approaches to digital health technologies.”
Please find the complete article here.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #medsyscon #regulatoryaffairs #MDUFA #SaMD #GSPR # IVD #IVDR
For further information please get in touch with us:
+49-176-57694801

05 Jan
GPT Applications in Healthcare?
Considering the possibilities and pitfalls of Generative Pre-trained Transformer 3 (GPT-3) in healthcare delivery
Diane M. Korngiebel, Hastings CenterGarrison, New York and Sean D. Mooney, Department of Biomedical Informatics and Medical Education, University of Washington Seattle, Seattle, wrote the following article published at https://www.nature.com:
“Natural language computer applications are becoming increasingly sophisticated and, with the recent release of Generative Pre-trained Transformer 3, they could be deployed in healthcare-related contexts that have historically comprised human-to-human interaction. However, for GPT-3 and similar applications to be considered for use in health-related contexts, possibilities and pitfalls need thoughtful exploration. In this article, we briefly introduce some opportunities and cautions that would accompany advanced Natural Language Processing applications deployed in eHealth.
A seemingly sophisticated artificial intelligence, OpenAI’s Generative Pre-trained Transformer 3, or GPT-3, developed using computer-based processing of huge amounts of publicly available textual data (natural language)1, may be coming to a healthcare clinic (or eHealth application) near you. This may sound fantastical, but not too long ago so did a powerful computer so tiny it could fit in the palm of your hand. GPT-3 and other technologies are getting close to passing a Turing Test, an assessment of whether the language such applications generate is indistinguishable from language produced by humans2,3. This possibility has generated both excitement and caution4, and Microsoft Corporation recently acquired an exclusive license from OpenAI for GPT-35. As with so many technologies and their potential use in eHealth, there are developments and applications that are unrealistic, realistic, and realistic but challenging—and perhaps unwise.
Natural Language Processing (NLP) has a long history in clinical informatics and includes groundbreaking work using computer-based algorithms that compute on text and natural language. There are many clinical applications of NLP including assisting with provider documentation, automated structured chart abstraction, and in machine learning6.
Despite the large amount of work in this area, AI that generates text and conversations, such as GPT-3, will not replace a conversation with another human being for the foreseeable future in clinical settings7. This means that it cannot interact with patients in lieu of healthcare providers or healthcare support personnel. Interactions with GPT-3 that look (or sound) like interactions with a living, breathing—and empathetic or sympathetic—human being are not8. A recent example of this failing was seen in testing the use of GTP-3 for mental health support using a simulated patient; the model supported the patient’s suggestion of suicide9. Moreover, language models such as GPT-3 are not grounded in input-diverse datasets (like visual and auditory data)1. GPT-3’s self-supervised prediction will, therefore, hit limits based on its pre-training data and cannot dynamically adjust a conversation or interaction for tone or body language.
GPT-3 is an autoregressive language model trained with 175 billion parameters and then tested in “few-shot learning settings” (in which a new language task can be performed after only a few examples). Autoregressive language models predict the next element in a text, usually a word, based on previous natural language texts. Although its developers at OpenAI think it performs well on translation, question answering, and cloze tasks (e.g., a fill-in-the-blank test to demonstrate comprehension of text by providing the missing words in a sentence)1, it does not always predict a correct string of words that are believable as a conversation. And once it has started a wrong prediction (ranging from a semantic mistake to using biased language), it does not go back and correct itself but continues to predict each word based on the preceding words. Further, since it is based on real language, human biases are present and, with inadequate priming of the application, may even be amplified and cause serious harm in sensitive contexts, such as healthcare. It is well-known that Internet-trained models reflect the scale of bias seen on the Internet, recently demonstrated by using the Implicit Association Test (IAT) to measure biases in a machine learning model trained on web-based content10. Therefore, it is unsurprising that GPT-3 showed associations between gender and certain jobs; often the default was male. Negative sentiments were associated with Black race and positive with Asian race. Islam was more often associated with terrorism-related words than were other religions1. Furthermore, according to recent research at the Center on Terrorism, Extremism, and Counterterrorism, GPT-3 is easy to prime for harmful text generation promoting extremism and terrorist activities, including Nazism and QAnon11.
It is within this caveat-filled context that evaluation of AI health and healthcare applications that produce natural language should assess their risk, feasibility, and return on investment—including prioritizing improved patient care. Realistic applications of GPT-3 must start in areas of high value, high feasibility, and low risk for all stakeholders, including (at a minimum) patients, clinicians, administrators, and payers. Applications with higher levels of risk or feasibility must be studied extensively and their actual and projected short-, medium-, and long-term impact measured. Realistic but challenging or unwise applications include those that are medium to high feasibility, medium to high risk, and medium to high value.
Unrealistic applications for GPT-3 applications in healthcare
GPT-3 is not an artificial general intelligence. It will not, and cannot (for now at least), replace a human interaction that requires humanness12,13. Although GPT-3 performed well on free-form conversation assessments demonstrating reading comprehension, it performed worst on a dataset meant to mimic the dynamic give-and-take of student-teacher interactions, and it also did not score well on multiple choice questions from middle and high school examinations1. This makes sense because it does not “know” anything. One of the major limitations of GPT-3 is that it repeats itself semantically, loses coherence over long conversations, and contradicts itself1,14. It would be unrealistic to consider GPT-3 as a stand-in for a healthcare provider or as a proxy in high-stakes interactions, such as a health emergency or an emotionally fraught interaction.
Realistic and feasible GPT-3 applications in healthcare
There is compelling promise and serious hype in AI applications that generate natural language. Some of that promise is realistic. Routinizing tedious work for providers could productively improve their work satisfaction and reduce time interacting with computer systems, a well-documented concern15. AI NLP applications could navigate complex electronic health record (EHR) systems, automate documentation with human review, prepare orders, or automate other routine tasks.
It is, however, capable of more complexity in its text conversations than a chatbot, including more natural-sounding question and answer exchanges14. This could personalize the experience of data collection in several non-critical healthcare system encounters, including online chat support for patients or assisting patients with setting up equipment in preparation for a telehealth visit. In fact, its developer, OpenAI, originally intended the software to be used by companies and organizations to improve customer service chatbots or do other similar tasks16.
However, each application must also include implementation guidance, including serious guardrails for all healthcare-related interactions. For example, this could mean it would be primed, perhaps using few-shot training alongside imposed limitations, to discuss solely relevant topics—and only after excisions of harmful, prejudicial, or inappropriate vocabulary.
Realistic but challenging GPT-3 applications in healthcare
Implementation guidance will be even more important in adapting GPT-3 technology for realistic but challenging healthcare applications. For example, using GPT-3 to assist with triaging non-critical patients presenting in the emergency department might seem a good use of the technology, from both a patient experience perspective and a resource allocation perspective. In this example, the focus would be on collecting accurate data from patients in a user-friendly way, thereby improving the patient experience (by making it easier to provide information) and enhancing patient care (by freeing up clinicians to spend more time in meaningful clinical encounters rather than routine data collection).
FDA approval would likely be required in this type of application in the United States and any evaluation must consider a broadly diverse population of patients. For instance, stakeholders, including developers and implementers, will need to be mindful of allocational and representational harms17,18, particularly when a virtual agent acts as a gatekeeper19—in which case the patient-user has no other option than to interact first with the virtual agent. In the triage example, the allocational harm occurs when those who are more able to successfully interact with the GPT-3 text intake process or forms are more likely to be triaged accurately. Implementation should include another means of triaging those patients who cannot, or do not wish to, use the conversational agent, which may also be too linguistically homogenous to offer culturally mindful language use. Furthermore, alternatives should be readily apparent and easy to access. Although this may seem to duplicate work, it is a necessary step in ensuring that harms are reduced and, as the GPT-3-driven interaction improves, redundant effort should be needed less often. An appropriate staff member would also need to review all triage forms completed using the GPT-3 application; it will be important to maintain a “human in the loop”. A representational harm in this triage example might be when the GPT-3 intake is only available in one language. In such a case, one could explore GPT-3’s expanded languages and translation functions: though the intake interaction could be in Spanish, form material could then be translated into the language spoken by the triage staff. There are real possibilities here, if done well, for language and related healthcare access barriers to be reduced. However, without careful implementation, including the step-wise process and oversight we describe, this triage application would be unwise with the potential to cause both immediate harm to individual patients and broader harm to patient groups, exacerbating healthcare disparities.
Making sure any realistic applications are equitable
AI software that generates natural language could be viewed as just another vendor-provided information technology tool, but it should not be. The role of human-computer interactions in informing the design of these conversational spaces, whether in an online chat or at an emergency department patient registration kiosk, will be critical to ensure not just that accurate and relevant data are collected, but also that the experience is what diverse users expect, want, and value. A broad range of stakeholders should be involved from the earliest stages of development (or tailoring) through deployment and evaluation. Stakeholders should be selected who can represent as comprehensive a view as possible on both the harms and benefits of proposed uses of GPT-3 in eHealth applications.
Transparency will be key to the appropriate use of GPT-3 types of technology. Human beings must be informed that the interaction is with a computer-based text generator. Doing so would address concerns that humans tend to anthropomorphize technology applications with human traits, assuming humanness and ascribing empathic emotional responses when there are none20,21. Some applications are highly feasible and seem low-risk but might harbor hidden hazards. For example, an innocuous natural language clinic appointment scheduler could not, with existing technology, detect a change of tone or social cues of nervousness a patient expresses and that might signal more urgent clinical needs.
Transparency is also critical for datasets and to disclose the limitations in language training activities. A GPT-3 application will need to be given conversation endpoints so that it leads the prompts rather than having the patient control the focus of the interaction; for form-completion tasks, it will also need additional guidance to determine whether the information a patient shares actually addresses the question posed. IT support personnel, or those in similar roles, will need to learn how to shape the prompts that will deliver the most relevant answers or results from a given interaction. For GPT-3 priming using few-shot learning, a commitment to transparency would require publishing any customized parameters. In high-risk applications in healthcare, including any patient-facing tools, such sharing must be mandatory.
We should have cautious optimism for the potential applications of sophisticated natural language processing applications to improve patient care. Additional concerns from our triage example include many implementation issues, including the ways AI software would interface with clinical and healthcare support workflows (a known concern for other AI applications22,23), how the data will be analyzed in real-time on the backend to successfully triage patients in a queue that prioritizes more concerning symptoms, and the degree and type of liability assigned the health system or provider. The future is coming. Rather than fear it, we should prepare for it—and prepare to benefit humanity using these applications. But for now, Dr. GPT-3 is not coming to a clinic near you anytime soon.
References
- Brown, T. B., et al. Language models are few-shot learners. Preprint at https://arxiv.org/abs/2005.14165 (2020).
- Turing, A. M. Computing machinery and intelligence. Mind LIX, 433–460 (1950).
- Lacker, K. Giving GPT-3 a turing test. Available at https://lacker.io/ai/2020/07/06/giving-gpt-3-a-turing-test.html (2020).
- Metz, C. Meet GPT-3. It has learned to code (and Blog and Argue). Available at https://www.nytimes.com/2020/11/24/science/artificial-intelligence-ai-gpt3.html (2020).
- Scott, K. Microsoft teams up with OpenAI to exclusively license GPT-3 language model. Available at https://blogs.microsoft.com/blog/2020/09/22/microsoft-teams-up-with-openai-to-exclusively-license-gpt-3-language-model/ (2020).
- Nadkarni, P. M., Ohno-Machado, L. & Chapman, W. W. Natural language processing: an introduction. J. Am. Med. Inform. Assoc. 18, 544–551 (2011).
- Warwick, K. & Shah, H. Passing the turing test does not mean the end of humanity. Cogn. Comput. 8, 409–419 (2016).
- Marcus, G. & Davis, E. GPT-3, Bloviator: OpenAI’s language generator has no idea what it’s talking about. Available at https://www.technologyreview.com/2020/08/22/1007539/gpt3-openai-language-generator-artificial-intelligence-ai-opinion/ (2020).
- Daws, R. Medical chatbot using OpenAI’s GPT-3 told a fake patient to kill themselves. Available at https://artificialintelligence-news.com/2020/10/28/medical-chatbot-openai-gpt3-patient-kill-themselves/ (2020).
- Caliskan, A., Bryson, J. J. & Narayanan, A. Semantics derived automatically from language corpora contain human-like biases. Science 356, 183–186 (2017).
- McGuffie, K. & Newhouse, A. The radicalization risks of GPT-3 and advanced neural language models. Preprint at https://arxiv.org/abs/2009.06807 (2020).
- Floridi, L. & Chiriatti, M. GPT-3: its nature, scope, limits, and consequences. Minds Machines 30, 681–694 (2020).
- Heaven, W. D. OpenAI’s new language generator GPT-3 is shockingly good—and completely mindless. Available at https://www.technologyreview.com/2020/07/20/1005454/openai-machine-learning-language-generator-gpt-3-nlp/ (2020).
- Elkins, K. & Chun, J. Can GPT-3 pass a writer’s Turing test. J. Cultural Analytics 2371, 4549 (2020).
- Sinsky, C. et al. Allocation of physician time in ambulatory practice: a time and motion study in 4 specialties. Ann. Intern. Med. 165, 753–760 (2016).
- LaGrandeur, K. How safe is our reliance on AI, and should we regulate it? AI Ethics, 1–7 https://link.springer.com/article/10.1007/s43681-020-00010-7#citeas. (2020).
- Abbasi, M., Friedler, S. A., Scheidegger, C. & Venkatasubramanian, S. Fairness in representation: quantifying stereotyping as a representational harm. in Proceedings of the 2019 SIAM International Conference on Data Mining (SDM) 801–809 (2019).
- Suresh, H. & Guttag, J. V. A framework for understanding unintended consequences of machine learning. Preprint available at https://arxiv.org/abs/1901.10002 (2019).
- Scherr, S., Haim, M. & Arendt, F. Equal access to online information? Google’s suicide-prevention disparities may amplify a global digital divide. N. Media Soc. 21, 562–582 (2019).
- Damiano, L. & Dumouchel, P. Anthropomorphism in human-robot co-evolution. Front. Psychol. 9, 468 (2018).
- Hortensius, R. & Cross, E. S. From automata to animate beings: the scope and limits of attributing socialness to artificial agents. Ann. N. Y. Acad. Sci. 1426, 93–110 (2018).
- Serag, A. et al. Translational AI and deep learning in diagnostic pathology. Front. Med. 6, 185 (2019).
- Kotter, E. & Ranschaert, E. Challenges and solutions for introducing artificial intelligence (AI) in daily clinical workflow. Eur. Radiol. 31, 5–7 (2021).”
Please find the complete article here.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #medsyscon #regulatoryaffairs #OpenAI #GPT #GPT-3
For further information please get in touch with us:
+49-176-57694801

19 Dec
IVD Software as a Medical Device (SaMD)
IVD Software as a Medical Device (SaMD)
Marcelo Trevino, global vice president, regulatory affairs and quality assurance at Agendia, a molecular diagnostics company focused on breast cancer genomic testing wrote in the current issue of meddeviceonline: “In recent years, software has been playing an increasingly critical role in the medical device world. Software with a medical intended use can be embedded as part of a medical device (and regulated as part of the device) or it can also be considered a medical device by itself and regulated as an independent device, which is called Software as a Medical Device (SaMD). In vitro diagnostic devices are known as IVD SaMD. This article summarizes the regulatory requirements in the EU, which are governed by the In Vitro Diagnostics Regulation (IVDR), and the requirements in the U.S., which are governed by the FDA under different parts of the Code of Federal Regulations and guidance documents, to allow manufacturers and developers understand the steps they must undertake to be able to commercialize these types of devices.
EN 62304:2006/A1:2016 is the standard on the life cycle processes for medical device software and it is considered the state of the art in medical device software development. It is recognized by FDA and IVD regulators as a consensus standard. The standard sets out requirements for the software life cycle, from development to maintenance of medical device software when software is itself a medical device and when software is an embedded or integral part of the final medical device. It also provides a framework of processes, activities, and tasks necessary in the software life cycle: a) software development process, b) software maintenance process, c) software risk management process, d) software configuration management process, and e) software problem resolution process.
The standard classifies software into three classes:
- A: No injury or damage to health is possible (requirements for software system shall be established during design and development);
- B: Non-serious injury is possible (requirements for software system shall be established during design and development and architecture of software shall be described); and
- C: Death or serious injury is possible (requirements for software system shall be established during design and development, architecture of software shall be described, and a description of the detailed design is required).
Once the initial safety classification has been carried out for the system, it is possible to break the system down into software items and software units that shall also be aligned with the design requirements.
A summary of the effects of the software classification on documentation and processes according to 62304 is provided below:
U.S. FDA Guidance
The FDA has published multiple guidance documents regarding the regulation of software, including SaMD. Some types of software are regulated as medical devices, whereas other types of software are not regulated.
There have been many digital health guidance documents published by FDA in recent years. Below are a few examples that can significantly aid manufacturers and software developers:
- Multiple Function Device Products: Policy and Considerations
- Content of Premarket Submissions for Device Software Functions (Draft)
- Digital Health Technologies for Remote Data Acquisition in Clinical Investigations (Draft)
- Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions (Draft)
- Changes to Existing Medical Software Policies resulting from Section 3060 of the 21st Century Cures Act
- Policy for Device Software Functions and Mobile Medical Applications
- Off the shelf software use in Medical Devices
- Clinical Decision Support Software
- Content of Premarket Submissions for Management of Cybersecurity in Medical Devices
- Medical Device Data Systems, Medical Image Storage Devices and Medical Image Communications Devices
- General Wellness: Policy for Low-Risk Devices
- Software as a Medical Device (SaMD): Clinical Evaluation
- Medical Device Accessories – Describing Accessories and Classification Pathways
- Deciding When to Submit a 510(k) for a Software Change to an Existing Device
- Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices
FDA has developed an interactive tool, called Digital Health Policy Navigator, to introduce digital health policies to stakeholders in an interactive format with simplified questions and yes and no answers. Manufacturers can assess if a particular software function meets the device definition and if it is likely it will be regulated by FDA as a device. The tool introduces policy considerations in plain language to allow manufacturers to easily understand policies and pathways.
As an additional resource, FDA recently published a list of currently marketed Artificial Intelligence (AI)/ Machine Learning (ML) Enabled Medical Devices. This list is a non-exhaustive list based on publicly available information that is meant to be a public resource on these devices and FDA’s work in this area and to show how AI/ML is being used across medical disciplines.
IVD SaMD Software Qualification in the U.S.
Software that meets the definition of a medical device under section 201(h) of the FD&C Act is deemed a medical device. IVD medical devices are regulated under 21 CFR 809. 21. CFR 809.3 (a) states:
“In vitro diagnostic products are those reagents, instruments, and systems intended for use in the diagnosis or disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body. These products are devices defined in section 201(h) of the Federal Food, Drug, and Cosmetic Act (the act), and may also be biological products subject to section 351 of the Public Health Service Act.”
Since there are many software applications and different categories, different regulatory approaches could be applicable depending on the nature of the software. This is particularly relevant when it comes to clinical decision support. Software used to inform clinical management is not regulated as a medical device if it is intended for a healthcare professional to independently review and understand the basis of the software’s recommendation and the software does not perform signal or image acquisition, processing, or analysis.
In September 2022, FDA finalized guidance on Clinical Decision Support that focuses on explaining four criteria necessary for clinical decision support software to be considered non-device clinical decision support. If software meets all four of the following criteria, it would be considered non-device clinical decision support software and would not be regulated as a device:
- The software function does NOT acquire, process, or analyze medical images, signals, or patterns.
- The software function displays, analyzes, or prints medical information normally communicated between healthcare providers.
- The software function provides information and/or options to a healthcare provider rather than a specific output or directive.
- The software function provides the basis of the recommendations so that the healthcare provider does not rely on any recommendations in order to make a decision.
EU IVDR
Regulation (EU) 2017/746 on in vitro diagnostic medical devices came into force on May 26, 2022. The regulation includes several types of software used in in vitro diagnostics and provides safety- and performance-specific performance requirements that manufacturers must comply with. The EU does not use the term Software as a Medical device (SaMD). Instead, the term medical device software (MDSW) is used for classification purposes.
IVDR SaMD Software Qualification And Defining Software Intended Use
The first step to qualify a software as an in vitro diagnostic medical device is to verify that the software is intended for in vitro diagnosis. According to Article 2 of Regulation (EU) 2017/746 on in vitro diagnostic medical devices,
“in vitro diagnostic medical device” means any medical device which is a reagent, […] software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens from the human body […] for the purpose of providing information on one or more of the following:
1. Concerning a physiological or pathological process or state;
2. Concerning congenital physical or mental impairments;
3. Concerning the predisposition to a medical condition or disease;
4. To determine the safety and compatibility with potential recipients;
5. To predict treatment response or reactions;
6. To define or monitor therapeutic measures.“
Software intended to be an accessory to in vitro diagnostic medical devices also falls under this definition and is subject to the same rules for demonstration of compliance. Manufacturers should then determine if the software creates information based on data obtained by IVD medical devices only. If it does, then the software is an IVD medical device, but if the data analyzed is obtained from an IVD and another medical device, then manufacturers must assess if the intended purpose is substantially driven by data sources coming from that other medical device since the EU MDR regulation could also be applicable.
MDCG 2019-11 provides examples of software qualified as in vitro diagnostic medical devices or that are excluded from the IVDR definition.
Software could be used in a healthcare environment but not considered an in vitro diagnostic medical device, such as, for example, software intended solely for communication, storage, or performing a simple search or software that supports the process from patient sample to patient result (such as laboratory information systems).
Software that combines and processes multiple in vitro diagnostic results (possibly combined with data from medical or non-medical devices) qualifies as an in vitro diagnostic medical device. For example, this category includes software that integrates genotype of multiple genes to predict risk of a disease or medical condition developing/recurring or software that uses an algorithm to characterize viral responses to different drugs based on genotyping assays.
There is also software that combines different modules, some intended for medical use and others that are not intended for medical use. According to the MDCG 2019-11, only modules intended for medical use are subject to the requirements of the regulation. It is the obligation of the manufacturer to identify the boundaries and the interfaces of the different modules based on the intended use. This will ensure that the combination of modules or systems is assessed for safety and does not impact performance of the modules subject to the medical device regulation as required per Annex I of the EU 2017/746 IVD Regulation.”
Please find the complete paper here.
For further information please get in touch with us:
+49-176-57694801

08 Dec
Time extension for MDR transition
In Short
The EU Commission will present elements of a legislative proposal for a targeted amendment of the MDR and IVDR during the EPSCO Health Council on 9 December 2022. Those elements are based on the input received so far from national experts and stakeholders and will include:
- an extension of the transitional period in Article 120(3) MDR with staggered deadlines depending on the risk class of the device. Those deadlines could be 2027 for class III and class IIb devices (i.e. devices with a higher risk) and 2028 for class IIa and class I devices (i.e. lower risk devices) that need the involvement of a notified body in the conformity assessment
- if needed for legal and practical reasons (including for access to third country markets), the extension of the transitional period could be combined with an extension of the validity of certificates issued under Council Directives 90/385/EEC and 93/42/EEC by amending Article 120(2) MDR
The Whole Story
The General Secretariat of the Council of the European Union published a paper “Implementation of the Medical Device Regulation – Information from the Commission” on 6 December 2022 which refers to an Annex covering an information note from the Commission on the subject “Any other business” at the meeting of the EPSCO Council (Health) on 9 December 2022:
“At the EPSCO Health Council on 14 June 2022, Health Ministers expressed their concerns that severe challenges related to the implementation of Regulation (EU) 2017/745 on medical devices (MDR) threaten the continued availability of certain medical devices needed for health systems and patients and may jeopardise the access of innovative medical devices to the EU market (1). Health Ministers called on the Medical Device Coordination Group (MDCG) to propose solutions to address, as a matter of urgency, immediate challenges related to insufficient capacity of notified bodies to certify medical devices in accordance with the MDR within the remaining transition period that ends on 26 May 2024 and the level of preparedness of manufacturers.
The Commission committed to report back to the EPSCO Council on 9 December 2022 and to present further proposals for solutions if needed.
1. State of play
At present, 36 notified bodies (2) are designated under the MDR. This is six more than on 14 June 2022. Further 26 applications for designation as notified body are currently being processed; three of them are in an advanced stage (3).
Based on the feedback received from notified bodies (4) to the latest survey in October 2022, notified bodies have received 8,120 applications from manufacturers and have issued 1,990 certificates under the MDR. According to a rough estimation presented by notified bodies to the MDCG on 17 November 2022, the number of MDR certificates issued by May 2024 may reach around 7,000 if the current rate of certificate issuance continues with no changes to current conditions. This is in stark contrast to 22,793 valid certificates issued under Council Directives 90/385/EEC and 93/42/EEC that will expire by 26 May 2024 at the latest. Of those 22,793 certificates, 1,387 certificates will have expired by the end of 2022, 4,311 certificates will expire in 2023 and 17,095 certificates in the first five months of 2024.
As regards Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR), the number of notified bodies designated remains low with only eight notified bodies designated so far; ten applications for designation are in progress, two of them in an advanced stage (5).
Pursuant to the data provided by notified bodies (6) during the survey in October 2022, notified bodies have received 822 applications from manufacturers and have issued 268 certificates under the IVDR. 1,551 valid certificates issued under Directive 98/79/EC will expire by 26 May 2025 at the latest. Of those 1,551 certificates, 38 certificates will have expired by the end of 2022, 165 certificates will expire in 2023, 482 certificates will expire in 2024 and 866 certificates in the first five months of 2025.
2. Actions undertaken since the June 2022 EPSCO Council
Following the calls expressed at the June EPSCO Council, the MDCG endorsed a position paper (MDCG 2022-14) on 25 August 2022, laying out 19 non-legislative actions within the current regulatory framework with a view to enhancing notified body capacity, access to notified bodies and manufacturers’ preparedness in order to support a successful transition to the MDR and IVDR.
Several of the actions listed in MDCG 2022-14 do not need further implementation, such as the status of guidance documents, or have already been implemented, such as a MDCG position paper on “hybrid audits” (7), and new MDCG guidance on appropriate surveillance with regard to devices covered by certificates according to the IVDD (8).
The MDCG has adopted a revision of MDCG 2019-6 with a view to removing obstacles to the employment of qualified personnel by notified bodies (9). On 26 October 2022, the MDCG taskforce on orphan devices held a stakeholder workshop to discuss a definition for ‘orphan’ devices and possible solutions for the regulatory challenges related to those devices. On 1 December 2022, the Commission adopted two delegated acts deferring the timing of the first complete re-assessment of notified bodies in order to free capacities both for designating authorities and notified bodies (10). Work on other actions is on-going, such as revision of the guidance on sampling of devices, on appropriate surveillance and on significant changes under the MDR, the provision of additional guidance regarding the practical application of clinical evidence requirements to legacy devices and the possibility for notified bodies to issue certificates subject to conditions, and clarification of what kind of dialogue between notified bodies and manufacturers before and during conformity assessment processes is permitted and should not be considered consultancy service. Some actions, namely those aimed to foster capacity-building of existing and new notified bodies, to facilitate access of small and medium-sized enterprises (SMEs) and first- time applicants to notified bodies and to increase preparedness of manufacturers, will be supported in the framework of the 2022 EU4Health Work Programme (11). While it does not seem to be feasible to quantify the exact impact of the actions laid out in the MDCG position paper 2022-14, the Commission considers that the message transmitted by the position paper, as well as the measures already taken are making a real difference. It is therefore important that the MDCG, with the support of the Commission, pursue its work to implement also the remaining actions.
At its extraordinary meeting on 17 November 2022, the MDCG agreed on a uniform and coherent approach to the use of market surveillance provisions as a bridging action before the transitional period would be extended by an amendment of the MDR. That approach is based on the application of Article 97(1) MDR by the competent authority of the Member State where the manufacturer is established in situations where a device is not in conformity with the MDR because its certificate issued under Directive 93/42/EEC or Directive 90/385/EEC expires before issuance of the necessary certificate(s) in accordance with the MDR. Application of Article 97(1) MDR will be subject to certain conditions and require the manufacturer to bring the non-compliance to an end within a reasonable period of time clearly defined by the competent authority. MDCG agreed to publish a paper describing that approach after the EPSCO Council meeting on 9 December 2022.
Further efforts are being made to support implementation in the longer-term through further actions (co-)funded under the EU4Health Programme. These include regular close monitoring of the market to get information on devices and certificates and launching reflections on how the MDR supports innovation, looking both at market aspects and governance (12) as well as supporting Members States in market surveillance activities through a Joint Action (13). The actions are expected to start in 2023.
3. Additional measures
Despite their full support for the MDCG position paper 2022-14, several Member States, some Members of the European Parliament and stakeholders consider that those actions will not be sufficient and are calling for additional measures, namely an urgent, targeted legislative initiative to amend the MDR. At the MDCG meeting on 24-25 October 2022, at the meeting of the Council Working Party for pharmaceuticals and medical devices on 28 October 2022, and at the extraordinary MDCG meeting dedicated to the transition to the MDR on 17 November 2022, a large number of Member States’ representatives spoke in favour of an extension of the transitional provisions provided for in Article 120 MDR linked to certain conditions in order to give manufacturers and notified bodies more time to conduct the necessary conformity assessment procedures. Notified body representatives, who participated in a part of the extraordinary MDCG meeting on 17 November, agreed on the need to give them and manufacturers more time to transition to the MDR. They indicated that an extension until May 2026 for class III devices might be too short for some of those high risk devices, in particular for those subject to consultation procedures. Notified body representatives also pointed out that notified bodies will need to spend a significant amount of resources in order to fulfil their duties under the MDR (e.g. surveillance of issued certificates; change notifications; review of manufacturer’s Periodic Summary Update Reports) that will increase the more MDR certificates are being issued. Hence, those resources would not be available for initial MDR certification.
The Commission has listened attentively to the concerns expressed by Member States, Members of the European Parliament and stakeholders, and, in light of the situation set out above, is ready to act upon these calls. The Commission will present the likely elements of a legislative proposal for a targeted amendment of the MDR and IVDR during the EPSCO Health Council on 9 December 2022.
Those likely elements are based on the input received so far from national experts and stakeholders and could include:
⮚ an extension of the transitional period in Article 120(3) MDR with staggered deadlines depending on the risk class of the device. Those deadlines could be 2027 for class III and class IIb devices (i.e. devices with a higher risk) and 2028 for class IIa and class I devices (i.e. lower risk devices) that need the involvement of a notified body in the conformity assessment;
⮚ if needed for legal and practical reasons (including for access to third country markets), the extension of the transitional period could be combined with an extension of the validity of certificates issued under Council Directives 90/385/EEC and 93/42/EEC by amending Article 120(2) MDR;
⮚ conditions to be fulfilled in order to ensure that the extension applies only to devices that do not present any unacceptable risk to health and safety, have not undergone significant changes in design or intended purpose and for which the manufacturers have already undertaken the necessary steps to launch the certification process under the MDR, such as adaptation of their quality management system to the MDR and submission and/or acceptance of the manufacturer’s application for conformity assessment by a notified body before a certain deadline (e.g. 26 May 2024);
⮚ the removal of the ‘sell off’ provision in Article 120(4) MDR and Article 110(4) IVDR.
Having regard to the urgent nature of the legislative initiative and the need for a swift adoption by the co-legislators in order for the changes to have the intended effect in a timely manner, the Commission considers that amendments should be kept to what is absolutely necessary at this moment in time.
Progressively, the Commission will also tackle more structural issues that have appeared, such as for instance those related to niche devices.
Please find the complete paper here.
For further information please get in touch with us:
+49-176-57694801

30 Nov
The promise of digital therapeutics
The promise of digital therapeutics
Austin Hackett, Amy Hung, Olivier Leclerc, and Sri Velamoor wrote a blog at https://www.mckinsey.com about the promise of digital therapeutics:
“Investments in digital-therapeutics companies in the United States have grown, on average, by 40 percent a year over the past seven years. What lies ahead for this fast-moving sector?
The Promise of Digital Therapeutics conference was hosted by McKinsey and Digital Therapeutics Alliance, a US-based not-for-profit trade association that aims to broaden the understanding and adoption of clinically evaluated digital therapeutics in healthcare. The conference brought together leaders and other representatives from pharmaceutical and digital-therapeutics companies, payers, and hospital systems to discuss the opportunities and challenges that lie ahead for this fast-developing industry. This article provides some of the key highlights from the conference and discussions on recent developments in digital therapeutics.
The pace of change
Investments in digital-therapeutics companies in the United States have grown by an average of 40 percent a year over the past seven years to reach more than $1 billion in 2018. Investors’ enthusiasm mirrors the growing demand for digital-therapeutic products and tools across the healthcare ecosystem—demand that two main trends have buoyed.
First is the technological development that is making available ever-greater amounts of data from which advanced analytics can mine insights. That has enabled the proliferation of personalized hardware, particularly smartphones and wearables. Second, and perhaps more important, is the growing body of evidence that digital interventions work. Peer-reviewed studies show improved outcomes from digital therapeutics, either alone or in conjunction with conventional protocols, in a broad range of indications, including cancer, ADHD, asthma, schizophrenia, and insomnia.
These trends are boosting the utility of digital therapeutics and hence interest in them from all stakeholders in the healthcare system. Patients, accustomed to consumer digital applications, want convenient and informative healthcare products. Physicians, many of whom are digital natives, are keen to manage their patients and businesses digitally. Payers and providers want digital tools that help them serve greater numbers of patients more effectively and at lower cost. Pharmaceutical and medical-device companies are seeking to develop digital solutions that improve current therapies and foster new ones. And the US Food and Drug Administration (FDA), keen to encourage digital innovation in healthcare, is evolving the regulatory framework accordingly.
A glance at some of the start-ups with products coming to market gives a sense of the variety of approaches to healthcare problems that these new digital therapies are utilizing. Products include video games to treat mental- and behavioral-health issues; a digital therapeutic platform that incorporates neurological music therapy, sensors, and artificial intelligence (AI) to help patients who have suffered a stroke or other neurological disorder to rebuild motor skills; and a smartphone app that can conduct electrocardiograms anytime, anywhere.
At the same time, large tech companies are combining their data-gathering and analytic capabilities with their vast scales to develop a new healthcare infrastructure. Amazon’s Alexa can diagnose health problems through simple voice commands, for example. Google is applying AI to many areas of healthcare. And Apple is making big advances in health through wearables that allow for continuous monitoring and the integration of electronic health records to enable patients to view all their data on their phones.
In sum, it is not hard to imagine the emergence of a very different healthcare system powered by digital technologies within ten years.
Regulation and approval for digital therapies
Digital therapies pose unique questions for the regulators charged with approving them. For example, the traditional quality-control measures for drugs, such as strength and purity, clearly do not apply. So what will replace them? What might an appropriate placebo be in clinical trials? What would a digital-therapeutic generic be? And would other elements on which the therapy might depend—perhaps the operating system, connectivity, or the device—also need regulating, and if so, how? Would, for example, a patient whose digital treatment depended on a good internet connection need an approved handset and internet provider?
The changing nature of digital-therapy products is also a consideration. How should regulators approve a product that, by design, will evolve continuously? As digital therapies learn through AI and machine learning, their algorithms will change, which means a product will no longer be the same as the one initially reviewed and approved. When should another review occur? And will regulators consider system improvements as formulations that can extend patent life—and so receive protection forever? Industry experts talk about digital products moving into a “perpetual Phase IV.” As new data accumulates, they will undergo testing against established standards, in ever-more-granular populations, and for new indications. This offers great potential in healthcare, but it also poses a new challenge for regulators.
The FDA, seen as a global leader in tackling the issues that digital therapies raise, is shaping regulation in digital health. It has been receptive to new proposals, to date, and is keen to introduce appropriate regulation. One of the best examples of this is the FDA’s Digital Health Software Precertification Program, a pilot for approving software-based medical devices. Conscious of how slow and ultimately unfeasible it would be to approve every software release for a digital product, the FDA uses this program to approve the developer instead. In this way, product development can occur efficiently through rapid iterations, not mired in a constant approval cycle. As of October 2019, the program included nine companies. More will join if the test phase is successful.
Although there are still many open questions, the participants in the Promise of Digital Therapeutics conference felt that the FDA was eager to work with industry to answer them. The message from executives at leading digital-therapy companies for newer companies in the industry was clear: “Engage with the regulators early and often. If you haven’t met with them yet, that should be the next thing you do.”
Digital-therapy adoption by consumers and healthcare providers
Digital therapeutics come in many varieties, each requiring different strategies to drive adoption. Nevertheless, those that succeed tend to offer three common features: meaningful incentives, human-centered design, and workflow integration.
Meaningful incentives
In a world in which consumers are inundated with information and approaches from companies wanting their attention, companies that offer meaningful incentives are often those that stand out, winning consumers’ engagement. McKinsey’s 2018 Consumer Health Insights Survey found that the innovative feature of insurance plans most appreciated by consumers was an incentive to change behavior. The majority said they would be willing to change their behaviors—exercise more, for example—to reduce their insurance premiums.
Not all incentives need to be financial. Gamification can be powerful too. A review of studies of digital-health applications showed that those with game-play elements helped improve motivation, engagement, and outcomes in treating arthritis, diabetes, back pain, obesity, and more. Evidence of positive clinical outcomes is another strong incentive that drives uptake and adherence to a therapeutic program.
Human-centered design
Digital-therapy companies must conduct rigorous and continuous user research to ensure that their product designs meet the needs and goals of patients and providers. Not enough do. McKinsey research has shown that users of digital therapies report lower satisfaction and less willingness to recommend the solution to other people than they do when using traditional healthcare products and services.
Likewise, consumers find their experiences with digital therapies lacking relative to the other digital products they use regularly, such as those offered by Amazon, Capital One, and Lyft. Each of those companies focuses on delivering an exceptional user experience based on extensive research, testing, and analytics. Each thinks deeply about the end-to-end experience and constantly evaluates improvements, such as product enhancements and integration with other software products. Digital-therapeutics companies should be aware that their products will be used at just one stage of a patient’s healthcare journey. They should therefore identify the other data and products that also form part of that journey and make sure their own products smoothly integrate with them.
Workflow integration
Any digital-therapeutic tool not carefully integrated into clinicians’ workflows will face significant barriers to adoption. For example, those that require clinicians to take additional steps to input more information will likely prove unpopular, as will those that require clinicians to shift in and out of different applications. So will those whose results do not integrate with existing patient data—although this issue might become less important as digital natives form a larger percentage of the workforce. By contrast, tools that help clinicians remove even small inefficiencies in their daily tasks will be welcome.
Companies must therefore engage clinicians early in the development process to understand protocols. Omada Health, a company that provides digital therapies used by several healthcare providers to manage chronic diseases, partnered with the American Medical Association to understand how to integrate therapies seamlessly with both clinical workflows and electronic health records.
Partnerships: Attraction and challenges
Successful digital-therapeutics companies have capabilities in areas that many biopharma companies traditionally do not: big data and advanced analytics, hardware engineering, human-centered product design, and innovative, flexible business models. Such capabilities will probably prove essential for pharmaceutical companies as healthcare evolves toward a digital future. But an acquisition to bring them in house would likely require significant investment and an appetite for risk, given the number of start-ups that fail. Hence the appeal of partnerships with digital-therapeutics companies instead. Pharmaceutical companies gain access to new capabilities and technologies, and digital-therapeutics companies benefit from greater scale and wider access to providers and patients.
Forging a successful partnership is hard, however. In a survey of digital-therapeutics-industry leaders, participants were asked, “On a scale of one to ten—with ten being “very difficult”—how challenging is it to drive a successful partnership between a digital-therapeutics start-up and a pharmaceutical company?” Eight was the most common response.
In sum, it is not hard to imagine the emergence of a very different healthcare system powered by digital technologies within ten years.
The panelists at the Promise of Digital Therapeutics conference shared their top tips, based on their experience, for how digital-therapeutics companies can ensure a successful partnership with a big pharmaceutical company.
- Ensure that operations and offerings can manage the scale-up that comes with a partnership.
- Do not overextend by trying to work with too many companies. Focus on managing a few partnerships well.
- Think of the user experience from the point of view of the partner, as well as of the customer, and design for both parties.
- Align incentives—so that, for example, both partners find the greatest value in the same sales channel.
The pharmaceutical companies that succeed in digital therapeutics may not turn out to be the most innovative, the most digitally capable, or the biggest investors in the industry. Instead, they may be those that are the best partners, striving to smooth the transition for digital start-ups that are working with a large pharmaceutical company for the first time. There are significant advantages to be gained by being the leader in “playing nicely with others.”
Who will pay? Reimbursement, value, and business models
The ultimate test of the value of a digital therapy is the amount a customer will pay for it. Among the factors considered, payers will typically value a therapy if given proof that it reduces healthcare costs, particularly by lowering acute-care utilization, reducing complications, or replacing expensive clinician visits with automated software or virtual visits. A proven ability to do any of these raises the likelihood of reimbursement for a digital therapy. Examples of companies attempting to demonstrate this are Myia Labs, whose product strives to reduce emergency hospital visits caused by coronary heart disease, and Pear Therapeutics, which aims to move care from the clinic to a cheaper setting.
The pharma companies that succeed in digital therapeutics may be those that are the best partners, striving to smooth the transition for digital startups that are working with a large pharma firm for the first time.
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

28 Nov
What’s Wrong With Your Medtech R&D Department?
What’s Wrong With Your Medtech R&D Department?
Larry Blankenship, director of Boulder iQ wrote in the current issue of www.meddeviceonline.com: “Voicing complaints about R&D departments in medical device companies is nothing new. Executives, staff members, and investors commonly ask, “What’s wrong with the R&D department? What’s taking so long? It takes them forever to get anything out!” Frustrated with continual delays, and seeing that R&D often spends most of its time supporting current device product lines, it’s not unusual for companies to look outside their organizations for innovative alternatives.
The reality is that R&D is indeed notoriously behind schedule and over budget, despite the introduction of countless tools and techniques to help organize and run such programs efficiently. In addition, R&D managers are serious, dedicated professionals. They certainly don’t intend to be late or over budget, and they generally are conscientious in estimating the resources they believe they’ll need.
So, what goes wrong? And what can be done? Here’s an overview of five common culprits in delaying or derailing device development in the R&D stage — and a solution that just may be R&D’s salvation.
1. (Dis)Similarity
Project management tools and approaches calculate the expected time and resources needed to implement a project, but they’re only as good as the data entered into them. That data often comes from comparing the current project to previous projects that appear to be similar. The problem is that the projects may not always be as similar as one assumes, and the comparative data may or may not fit accurately.
For example, a recent catheter project was to incorporate a novel profile-changing tip. The R&D team had developed many catheters before and saw this as a “similar” project. In design, though, they found that the techniques they had used for previous catheters would not work for this tip. Instead, they needed special tooling and techniques. The result: the project went over budget by 40%. If time had been designated early on to investigate and research the needs in more depth, the R&D team could have developed a realistic assessment of what was and was not “similar” and kept the project more on time and on budget.
2. No Recognition of Research Tasks
“Engineering tasks” are those that can be successfully implemented in a straightforward, predictable manner using methods known and previously applied. “Research tasks” are those that a company does not know exactly how to do at the moment and needs to figure out. Inability to recognize the difference, and thereby not accounting for the necessity of research tasks in planning and budgeting, will severely derail R&D plans.
One real-life example of an important hidden research project comes from the intravenous pump industry. Looking at an existing a standard device that used a 5 ml reciprocating syringe, a competitor decided to leapfrog the market by making a 0.5 ml pump. Their idea was that the patient could wear the device, versus having to deal with the device clamping onto an IV pole. It may have been a great idea, but to be competitive, the developer still had to demonstrate 2% accuracy and offer similar disposable pricing. In reducing the size by a factor of 10, though, the manufacturing tolerances on the syringe could no longer be met with standard molding approaches. The resulting cost increase took the device to the point where it was no longer competitive.
3. Feature Creep
Fundamental to projecting a development timeline and budget is a solid understanding of what the product is, what it does, and how it’s used. Early in the project, a product requirements document should detail all features that define these elements. Once development is underway, making any changes to that document — in effect, adding functions, or “features” — can be devastating to a planned timeline and budget.
For example, marketing may ask to add “just one more control knob on the front panel.” Once the control panel has been laid out, the circuit boards printed and parts received, this seemingly simple addition can be highly disruptive to the device development.
The point at which a feature change request is made is critical to its impact. Consider a request to add an inline injection port to a tubing set. If the tubing set has already completed eight weeks of biocompatibility testing, adding the injection port may require a complete re-test, which could mean another two months — or more — to the project timeline. Obtaining enough input up front to establish solid, unchanging product requirements and avoiding feature creep later in the program is essential to project predictability.
4. Time Distractions
Protecting project resources is an important task for an engineering manager. This includes establishing policies to keep the R&D team focused on the new product’s development, versus risking them being pulled away to deal with current devices issues. Importantly, it also means working with executive management to communicate the impact of such a diversion of resources.
To make this type of system work, some organizations have implemented a special engineering group, “manufacturing and sustaining engineering,” to address issues with devices already in production. This allows the R&D team to focus on new devices without diversion.
5. Corporate Culture
How management deals with issues and delays in R&D sets the tone for how well the department can address issues, now and in the future. It’s not uncommon for corporate executives to fire R&D managers out of frustration at delays. Those replacing them will ultimately face the same situation down the line unless the approach changes.
And, in a culture where R&D personnel fear for their jobs, they may start to inflate estimates of time and costs of project tasks to cover themselves. When such culture exists, the distrust is damaging to all parties involved.
The keys to success are to promote in-depth advanced thinking that addresses the problems described here, to work with the R&D team to plan with adequate forethought and discovery, and to execute with an eye toward predicting potential challenges and advanced preparation.
Phase 0 Solution
“Solution” may be an overused buzzword. But in this scenario, the good news is that there really is a solution to the R&D problems described. It’s called Phase 0.
Simply, put, Phase 0 is a phase that comes before the typical five product development phases most medical device companies use. Those five defined phases align with the required design controls of the FDA’s Quality System Regulation.
- Phase 1: User Needs
- Phase 2: Design Input
- Phase 3: Design Process
- Phase 4: Design Output
- Phase 5: Verification and Validation
Generally, each of these phases must be completed, then accepted by quality assurance, before the next phase begins, thus creating a “gate” of review and approval between each phase. While this phase-gate approach is easily auditable to show compliance with the Quality System Regulation, it is also inherently inefficient from a project flow perspective.
For example, the IFU is typically part of Phase 3 or 4, which take place when the design is largely complete. If a problem in the user interface comes up, it may require modifications to Phase 1 and Phase 2, which means going backward, not forward. Or, consider Phase 5, where verification and validation test protocols are written. The design team may not have been aware of the rigors of the testing or required test points, forcing some level of redesign.
Enter Phase 0. It involves preparatory work to determine if a device development project can even gain authorization. In fact, a main purpose of Phase 0 is to cancel development projects before they get off the ground.
Killing a project can be one of the toughest jobs for R&D managers (and company leadership). Beyond the fact that projects and devices can become so personal, no one wants to cancel a project after years of development and investment of millions of dollars. Phase 0, executed objectively, in line with sound business decisions, can eliminate emotional and subjective attachments to projects. The bottom line is that it’s much easier and cheaper to cancel a project after Phase 0, before it really gets started.
A Phase 0 document clearly states that the work is preliminary investigation and research that may or may not be used as the basis for initiating a formal device development project. It therefore is not subject to FDA design controls.
Phase 0 is not trivial — or short. It typically takes several months. Yet we have seen, over and over again, for more than a decade of implementing Phase 0 with clients, that it is a wise investment. Phase 0 activities are designed to uncover the unknowns, take question marks out of equations, and eliminate the age-old adage plaguing medical device companies: “We didn’t plan to fail, we failed to plan.”
Phase 0 Tasks
Since Phase 0 is not subject to design controls, a company can define the activities and deliverables in a way that best serves its needs and device attributes. Many device firms work with a product development consulting firm that has hard and fast experience in the implementation of Phase 0. Whether you go this route or do it yourself, tasks usually begin with concentrated focus on defining the users, their attributes and training, and their needs in function, environment, and process. Well-organized brainstorming sessions, which include knowledgeable people outside the primary device development team, bring in objective ideas and perspectives.
From there, the R&D team can perform an initial written feasibility assessment of the top concepts, with a list of likely engineering and research tasks. Moving forward with the top two or three concepts, the team can update the device requirements document, and, for each concept, create sketches/illustrations and generate an IFU and a perceived workflow diagram.
A prime concept will emerge from user input (following ISO 62366 Usability Engineering for a Formative Usability Study). R&D can take that concept to a demonstration level, create the IFU and workflow diagram for it, and finalize based on additional user feedback.
From there, it’s back to the engineering and research task list for updates and appropriate inclusion in the project plan. With the Phase 0 results, the plan — in whatever form it takes — should have buy-in from the R&D team and other corporate decision-makers. And if the project does move forward, good documentation of all Phase 0 investigations, research, tests, and activities will greatly streamline the formal design control project phases.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

23 Nov
Five ways that life science companies can build tech talent
Mike Joyce, partner in McKinsey’s Washington, DC, office; Jeffrey Lewis, senior partner in the New Jersey office, Suman Thareja; and Robert Machinek, associate partner in the Boston office wrote an article in the blog series “The rise of health technology” at www.mckinsey.com: “For life science companies to meet their digital ambitions, they must strengthen their technology skills. In a challenging market, the industry is uniquely positioned to shine.
The pace and urgency of digital transformations in life sciences have increased rapidly in the past two years. The COVID-19 pandemic has forced life science organizations to rethink their commercial models. The most obvious effect has been the shift to digital channels for interactions between healthcare providers and patients as face-to-face contact became more problematic. In addition, the pandemic has created supply chain uncertainties that made data-driven operations and planning more critical. And clinical trials have necessarily become increasingly decentralized, virtual, and customer centric.
These changes have dramatically raised the profile and importance of technology in the industry. Accordingly, there is a new sense of urgency in hiring and retaining the talent required by these changes.
Life science organizations should have little difficulty attracting and retaining tech talent. The salaries that they offer are typically higher than those in other industries. The work—improving health and the quality of life—is compelling, as is the industry’s core mission to innovate.
Despite this, most life science organizations struggle to find and hire the needed talent—including full-stack engineers, cloud architects, and site reliability engineers—to scale and sustain their digital transformations. Several macrotrends are responsible for this difficulty:
- Digital tech is an increasingly critical enabler of scientific breakthroughs. The technology is vital for remote clinical trials, biomarker identification, and digital therapeutics.
- Tech capabilities are rapidly evolving. As the pace of tech innovation accelerates, the roles and skills needed to enable differentiating tech solutions, products, and services are quickly changing. Organizations with traditional approaches to hiring, upskilling, and reskilling are struggling to keep up, as they often fail to identify the talent they need or fully understand their expectations and requirements.
- The Great Resignation is real. Many employees are leaving their jobs, largely because of a misalignment between what they seek and what employers provide. This has widened the talent demand and supply gap.
- The competition for tech talent is industry—and increasingly, location—agnostic. While for many critical roles, life science organizations compete only with one another, every industry is fishing in the same pool for tech talent. Much of the work can be performed remotely, which has eliminated regional advantages and made competition fiercer.
While for many critical roles, life science organizations compete only with one another, every industry is fishing in the same pool for tech talent.
Building tech talent with life sciences’ unique strengths
Based on our experience in helping life science organizations with tech talent transformations, we believe the industry can find the right talent by leveraging its unique strengths. Some life science organizations today are already succeeding by radically transforming their organizational structures, operating models, and HR processes.
We have learned from those leading organizations that it’s important to engage with tech talent across the entire hire-to-retire life cycle. Based on our observations of their successes, we have identified five actions that CEOs, chief information officers, chief HR officers, and functional leaders in life science organizations can take to build tech talent.
1. Establish skill-based, strategic workforce planning
The skill sets that life science companies needed three years ago are different from the ones that they need today. Many companies are moving away from traditional tech delivery models (with throngs of project and relationship managers) toward more agile models (with product roles, designers, and internal engineers). New, cloud-based architecture requires different engineering skills from before. And as innovative, open-source tools and languages are adopted, updated skill sets are needed.
Strategic workforce planning allows organizations to take a proactive approach to refreshing their tech talent bench. Many life science organizations have been reactive, plugging talent gaps as they emerge by outsourcing to vendors, which is often unsustainably expensive. Plus, tech talent is increasingly a strategic differentiator. Digital business advantages can’t be achieved by outsourcing to vendors, which historically have struggled to provide innovation.
A strategic approach requires aligning talent planning with a tech–business road map. For instance, comparing a company’s tech talent with what will be needed for its projected product pipeline allows the company to hire, upskill, reskill, and partner in a more targeted manner.
One global pharmaceutical company based in the United States recently implemented a skill-based approach to tech workforce planning as part of a broader shift to product management and greater agility. It upskilled legacy project managers into scrum masters, determined the redeployment needs of its automation teams, and developed hiring goals for new product owner roles. The outcome was an optimal design of 20 pilot squads comprising 150 cross-functional members with clear, skill-driven roles. Furthermore, the company’s IT organization gained new insight into its labor costs, allowing it to invest more in a new product-centric model.
2. Have an authentic, digitally focused value proposition
Our research has shown that having a clear employee value proposition (EVP) can make a company more attractive to potential employees. Life science organizations, along with emphasizing their commitment to patient outcomes, have largely focused their value proposition on employees in R&D and commercialization—that is, those who traditionally returned the greatest benefit to the business. However, we believe that to compete successfully for tech workers, life science organizations can do better with a digital-specific EVP (see sidebar “How to differentiate your life science company’s digital brand through an employee value proposition”).
One European pharma company established a digital center of excellence. It took its vision for the new unit to market, publicly communicating the center’s strategic importance and demonstrating to applicants its place at the organizational and geographic hearts of the company. The company’s website for tech applicants shows the core tech products that it is developing, with success metrics that are similar to those for its traditional products. The company also quotes healthcare providers and business stakeholders who attest to the impact of its digital products. For a tech solution related to neurodegenerative disease, for example, it describes how the tech can help patients, establishing a direct link between tech innovation and better healthcare. These EVP improvements have enabled the company to choose the best candidates from a large pool of applicants and quickly scale up its digital center of excellence.
What also excites and attracts tech talent is the technology that a company uses. Organizations can identify the most appealing technology by analyzing online job boards and monitoring employee reviews. Additionally, it’s crucial to represent the organization accurately. Otherwise, companies risk attrition when new hires realize that the EVP (a de facto social contract) doesn’t align with reality. Organizations should be honest in setting expectations, whether the company is ramping up its technology or is well along in a large-scale digital transformation. It is also critical to communicate how the current state will benefit the tech workforce. For example, joining at the start of a life science organization’s digital journey provides opportunities to see projects through from beginning to end, something typically reserved for more senior personnel in more technologically mature organizations.
3. Take a candidate-centric approach to hiring
People live in a consciously facilitated, frictionless, experience-based world, from ordering coffee to paying a friend on a digital payment app to standing in line at Disney World. A job seeker shouldn’t experience finding their next job or role as painful or complicated.
Like any other customer experience, the hiring process can benefit from design thinking—a problem-solving approach that prioritizes consumer needs in improving products and services. Companies can improve their design thinking by defining the current process from the vantage points of new hires, hiring managers, recruiters, and online reviewers to find out what works well and what doesn’t. Once a company has established the baseline, it can convene stakeholders to design an optimal hiring experience.
One approach is to build out personas of internal and external candidates—who they are, what motivates them, and what their career aspirations are. A company then creates an efficient process that removes burdensome steps by streamlining how candidates are screened and assessed for technical and behavioral skills (online skill assessment tools can accelerate this process).
This can provide a competitive advantage when candidates compare their experiences in various hiring organizations (see sidebar “The hiring journey: Where will you create moments of delight for a candidate?”).
One inspiration that the life science industry can draw from is how it has adopted new customer engagement models and improved patient and healthcare provider experiences in response to the COVID-19 pandemic. Many life science organizations have all the capabilities needed to create compelling journeys and experiences but haven’t yet directed them toward hiring tech talent.
Many life science organizations have all the capabilities needed to create compelling journeys and experiences but haven’t yet directed them toward hiring tech talent.
One Japan-based pharma company used journey mapping to reinvent its talent onboarding. It established a consistent set of metrics to track the efficacy of the new process, the quality of the hires, and the effectiveness of the hiring manager and teams. The resulting data enabled the organization to make step-change improvements.
4. Embrace agile ways of working
Tech staff today expect to participate in small, nimble teams within the framework of a flexible and agile organizational model. Such approaches are becoming more common for life sciences R&D teams, such as bench scientists and clinical trial leads.
For tech talent, that model means minimizing command-and-control processes and allowing team leaders to focus on encouraging their teams, providing guidance on interpreting road maps, and enabling the path forward—in other words, embracing agile ways of working on the day-to-day level. Teams self-select their focuses, test and release products iteratively, and refine their working models and interactions with each sprint. This often translates to tighter integration among tech teams, the business, and customers, allowing tech talent to contribute to an organization’s health- and patient-centric mission rather than work on abstract tech requirements handed to them at arm’s length.
For example, Johnson & Johnson champions an operating model that enables high-performing, cross-functional, autonomous teams to work continuously on specific products. The results have included improved employee satisfaction, a reduced cost to the business for delivery, increased speed, and a reduction in non-value-added work.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

10 Nov
FDA’s New Draft Guidance on Cybersecurity
FDA’s New Draft Guidance on Cybersecurity
Brynn Stanley, J.D., Associate Attorney, Gardener LAW, wrote in the current issue of gardner.law: “The FDA has been continuing to work on protecting medical devices from the threats of cybersecurity. In April of this year, the Agency released the latest draft guidance addressing cybersecurity in the medical device lifecycle. There are several major changes in the 2022 version, which replaces the 2018 draft guidance.
Gardner Law recently presented a CLE presentation (click here to view) on the topic of cybersecurity that outlines:
- Background on the cybersecurity and the FDA’s approach
- Major changes from the 2018 draft guidance
- Highlights of the 2022 release
No time to watch the video? What follows is a brief summary of the video presentation.
Background on cybersecurity and the FDA’s approach
The FDA has been continuing to strengthen and evolve its approach to addressing cybersecurity in medical devices. As noted in the April 8th Federal Register Notice, “[t]he need for effective cybersecurity to reasonably ensure medical device safety and effectiveness has become more important with the increasing use of wireless, internet- and network- connected devices, portable media (e.g., USB or CD), and the frequent electronic exchange of medical device-related health information. In addition, cybersecurity threats to the healthcare sector have become more frequent, more severe, and carry increased potential for clinical impact.”
The 2022 draft guidance emphasizes the importance of ensuring that devices are designed securely and are capable of mitigating emerging cybersecurity risks throughout the Total Product Life Cycle (“TPLC”). This change to a TPLC approach will impact manufacturers as they begin to incorporate these concepts into not only their premarket submissions, as was the focus of the 2018 draft guidance, but also into their Design Controls processes. In fact, effects of the TPLC approach will be felt throughout a manufacturer’s Quality Management System (“QMS”). This is likely to have a big impact on manufacturers, especially for those with legacy products or for companies playing “catch-up” to the 2018 draft guidance.
What has changed since 2018?
As was mentioned above, the biggest and most impactful change to manufacturers is the TPLC approach to cybersecurity, but there were several other changes implemented:
- Risk Tiers have been removed. The concept of risk has been preserved, however.
- Cybersecurity Bill of Materials (“CBOM”) has been replaced with Software Bill of Materials (“SBOM”). This was done to reduce some of the burden on manufacturers when the vast majority of cybersecurity concerns are within the software, itself, and not the hardware. The SBOM concept also aligns the draft guidance more closely with the May 2021 Executive Order on Improving the Nation’s Cybersecurity.
- Additional clarification regarding premarket submission document requirements has been added throughout the draft guidance.
- Investigational Device Exemptions (“IDEs”) have been added to the scope.
- A change in title was made to better capture the scope of the current draft guidance: Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions Draft Guidance for Industry and Food and Drug Administration Staff.
- A document structure change was made to align with use of a Secure Product Development Framework (discussed further below).
What is included in the 2022 draft guidance?
Secure Product Development Framework
A Secure Product Development Framework (“SPDF”) is a set of processes implemented by the device maker designed to mitigate the number and severity of vulnerabilities in products throughout the device lifecycle.
The FDA proposes using a SPDF as a means of satisfying a manufacturer’s Quality System Regulation (“QSR”) obligations and including it as part of the product development process. A SPDF should be deployed throughout the TPLC.
Risk Management
For the 2022 draft guidance, the FDA has greatly expanded their expectations around risk management. While manufacturers have been incorporating aspects of risk management within their QMS and product development processes for some time now, those processes may not be robust or sophisticated enough to fully assess the risks for cybersecurity under the latest draft guidance.
The 2022 draft guidance focuses heavily on the use of “Threat Modeling” as part of risk management for cybersecurity. Threat Modeling identifies security objectives, risk, and vulnerabilities; defines risk controls or countermeasures to mitigate risks; and supports risk analysis activities. Manufacturers will need to reassess and likely change their approach to risk management to ensure compliance. More information on Threat Modeling can be found in the Playbook for Threat Modeling Medical Devices, which was commissioned by the FDA and released in November of 2021.
Cybersecurity and Premarket Submissions
The FDA assesses the adequacy of device security based on the ability of a device to provide and implement the security objectives of authenticity, which includes: integrity, authorization, availability, confidentiality, and secure and timely updatability and patchability.
Premarket submissions should address how the above security objectives are met. Another thing manufacturers need to consider in their premarket submissions is substantial equivalence, as the FDA is now planning on taking cybersecurity controls into consideration in making a substantial equivalence determination. Again, moving forward, manufacturers will want to look at their current systems, to make sure cybersecurity is considered early and often to avoid delays once they reach the point of submission.
To be effective, cybersecurity needs to be “baked in” and not “bolted on”. Manufacturers will need to make changes to their approach. Regardless, we recommend manufacturers start planning and implementing the concepts found in the draft guidance now, in both their products and within their QMS, in order to ensure they maintain compliance.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

26 Oct
Tips For Your Virtual Meetings With The FDA
Kate Cook, Catherine M. Cook Group LLC, is an FDA veteran who has worked in legal and policy positions in the Office of Chief Counsel, as well as in the FDA’s CBER and CDRH, where she served as associate director for regulations and policy and wrote in the current issue of meddeviceonline: “In the past two years, the pandemic has kept sponsors developing drugs, biological products, and medical devices from meeting in person with FDA. Advisory committee meetings, workshops, and conferences have gone virtual, too. While some sponsors have hopes that changing pandemic conditions will open the doors to in-person meetings, this seems unlikely.
In-person meetings with FDA officials are likely to remain the rare exception, not the norm. The FDA commitment letter developed in the latest round of prescription drug user fee negotiations sets out FDA’s commitments to meet “face to face” with the sponsor at specified stages in a product development program, but defines “face-to-face” to “include… both in-person meetings and virtual meetings on IT platforms that allow for both audio and visual communication.”
This means that FDA can opt to meet virtually instead of in person, and it has strong reasons to do so. Meeting virtually avoids the administrative work needed to bring outsiders onto the secure FDA campus, and the format more easily accommodates individual employee schedules. There are very clear scheduling advantages to virtual advisory committee meetings, which are easier to schedule when busy experts are not required to spend a half day or more traveling to and from an in-person meeting.
Moreover, it can seem impossible to bring FDA meeting participants together in one conference room. FDA staff are increasingly likely to be working remotely at locations nowhere near the FDA’s White Oak campus in Maryland. FDA officials have made recent statements about the value in authorizing remote work as a recruitment tool, and it is a tool FDA is likely to be using.
FDA has a lot of recruiting to do. In the latest round of user fee agreements, the agency committed to significant numbers of targeted hires to support the prescription drug user fee program, including, for Fiscal Year 2023, 132 CBER staffers and 77 CDER staffers. Recruiting qualified scientific employees is always a challenge for FDA, but it will also need to address recent staff departures. Exhausted by the work of developing and supporting the FDA’s pandemic response, experienced staff members are retiring or seeking outside employment in significant numbers. This exacerbates existing understaffing and can lead to significant losses of expertise and institutional memory.
Once these hundreds of new employees are hired, FDA will have to develop employees who work remotely and do not have the benefit of daily in-person interactions with their managers and mentors — who may also be working remotely. This will present management and supervisory challenges for the agency and may change its work culture significantly. One thing for sponsors to look out for is that new reviewers sometimes default to an overly conservative position. Sometimes, it may be necessary for sponsors to ensure that supervisors are involved in giving FDA advice, particularly on novel issues.
Obtain FDA Advice By Submitting A Meeting Request
The primary mechanism for sponsors to obtain advice from FDA on their development programs is by filing a request for a meeting. When FDA grants the meeting, it may be a “face-to-face” meeting (defined to include a virtual meeting), a teleconference, or a written response only. FDA and industry have described different meeting types in user fee commitment letters for prescription drugs and biological products (PDUFA), biosimilars (BsUFA), generic drugs (GDUFA), and medical devices (MDUFA) as well as in related guidance documents addressing meetings under PDUFA, BsUFA, GDUFA, and MDUFA.
These guidance documents are likely to be revised to incorporate new user fee commitments under the recently concluded negotiations, once Congress has enacted the legislation necessary to reauthorize these user fee programs. For example, the new PDUFA commitment letter addresses two types of meetings for the first time: INTERACT meetings and Type D meetings. An INTERACT meeting provides an opportunity for sponsors to discuss novel questions and unique challenges in early development, prior to filing an IND. A Type D meeting is focused on a narrow set of issues (typically not more than two issues and associated questions). Requests could include a follow-up question that raises a new issue after a formal meeting, a narrow issue on which the sponsor is seeking agency input with only a few associated questions, or a general question about an innovative development approach that does not require extensive, detailed advice.
FDA’s commitment letters and guidance documents describe the criteria the sponsor must meet to be granted a specific type of meeting. Although FDA’s user fee commitments recognize a right to a face-to-face meeting at certain stages in a product development program, under other circumstances FDA may decline to schedule a face-to-face meeting and will provide a written response only. The commitment letters and guidance documents address the timeframes for the sponsor to submit their briefing document and for FDA to respond to requests and to schedule meetings.
FDA has committed to send preliminary written responses to a sponsor’s questions contained in their background package. When a meeting has been scheduled, FDA will send the response in advance of the meeting, giving the sponsor the opportunity to focus the meeting on discussing certain FDA responses, or even to determine that the written response is adequate and a meeting is no longer necessary.
Optimize The Timing Of Your First Meeting
Some sponsors want to initiate meetings with FDA as early in development as possible. FDA has recognized that emerging technologies raising cross-cutting issues may warrant very early discussion and has created forums for such discussions in the Critical Path Innovation Meeting, CDER’s Emerging Technology Team Meeting, and the CBER Advanced Technology Team Meeting. However, a sponsor that obtains advice too early in preclinical development of a specific product may find that significant pre-IND questions arise a short time after a pre-IND meeting because they decided to modify the product in important ways. Since FDA generally limits a sponsor to one pre-IND meeting, the sponsor may have to ask for advice on their new questions using a different meeting request format, such as a Type C meeting, which has longer timelines for agency response, or the new Type D meeting if FDA agrees that the scope is narrow enough for such a meeting. FDA has advised that for products regulated by CBER’s Office of Tissues and Advanced Therapies (responsible for regulating cell and gene therapies, among other biological products), a pre-IND meeting is appropriate if:
- The sponsor has defined the manufacturing process to be used for the clinical studies and has developed assays and preliminary lot release criteria.
- The sponsor has completed proof of concept (POC) and possibly some preliminary preclinical safety/toxicology studies and desires to move to the definitive toxicology studies.
- The sponsor’s questions involve IND-enabling chemistry, manufacturing, and controls (CMC), pharmacology/toxicology (P/T), and/or clinical trial design issues.
Focus On Improving The Video/Teleconference Experience
A sponsor should assure that all sponsor attendees can access the video conference platform successfully, and that audio is clear and bug-free. The sponsor’s meeting plans should address not only who is responsible for the sponsor’s presentation, but also who is the critical note taker and who is authorized to call for a short internal conference if needed during the meeting. The sponsor should be able to mute the sound to FDA in case there is a need to hold a sidebar with sponsor attendees only.
If possible, all sponsor attendees should assemble at the same location. This can be challenging, given that sponsor employees often work at different locations and that a sponsor’s external consultants may be in attendance, but having everyone in the same room can allow a sponsor to adjust strategy in real time after an internal sidebar discussion. If it is necessary for sponsor representatives to participate from multiple locations, participants should have a plan in place to address the problems that can occur when one site loses connectivity. This may include identifying and preparing back-up presenters for critical presentations.
In addition, the sponsor should think about how their representatives are likely to present themselves to FDA and be on the lookout for presenters who may come across as dismissive of FDA questions and concerns.
Make Your Case And Request Participation By Appropriate FDA Experts
A sponsor’s briefing book should make its case in support of the sponsor’s position on the issues they are asking FDA to advise on. This is especially important for a sponsor seeking FDA support for an innovative or less burdensome approach. If specific subject matter expertise is important to understanding the issues, a sponsor should request the participation of specific FDA employees or representatives of specific offices. For example, if an issue requires CMC or other manufacturing expertise, the sponsor may request the involvement of a representative from CDER’s Office of Pharmaceutical Quality or CBER’s Office of Compliance and Biologics Quality.
Value Your Project Manager
A sponsor’s primary point of contact with FDA is the project manager assigned to the development program. Sponsors’ regulatory affairs personnel generally understand the value of a good relationship with the project manager, who can sometimes work magic in facilitating informal communications between the sponsor and regulators.
Understand What Your Regulators Are Focusing On
FDA’s written response to a briefing package may identify issues of concern to FDA. The sponsor should take every opportunity to probe the scope and significance of these concerns in the meeting and in future product development discussions. It sometimes happens that FDA raises an issue in early correspondence with a sponsor whose application for approval, filed years later, is not successful due to the same issue raised in early communications. Sometimes the early notice has prepared the sponsor for this possibility, and the sponsor is ready to make its case for approval on appeal or has already initiated studies to develop data to satisfy FDA’s concern. However, sometimes the sponsor will have discounted the significance of FDA’s early comments and is unprepared to address them.
A sponsor should also pay attention to the FDA staff attending the meeting. If you are unfamiliar with someone and their FDA role, do a little research. You may learn from an attendee’s slide presentations that they specialize in a specific issue, such as the distribution of a drug under a Risk Evaluation and Mitigation Strategy (REMS) program. This may signal that the parameters of a REMS distribution program for your drug are the subject of discussion within FDA. Similarly, the involvement of other individuals or offices may signal that FDA is focusing on a drug exclusivity or other type of issue.
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

21 Oct
Medtech Cyber-Incidents – A Costlier Problem Than You Think
Elliot Turrini, CEO of Practical Cyber and Ben Locwin, a healthcare futurist and medtech executive wrote in the current issue of meddeviceonline:
Harnessing advanced computing technologies — e.g., “artificial intelligence (AI), faster and smaller computing hardware, computing-driven robotics, augmented/virtual reality, and Internet of Things (IoT)” — gives medtech companies almost unlimited profit opportunities, as our May 4, 2022, article explained.
Our article warned that this opportunity comes with “almost unlimited liability” from cyber-attacks. While medtech companies have heard general cyber-attack warnings like this for years, many are still woefully underprepared — particularly because they materially underappreciate the financial danger of cyber-attacks. This happens largely because of the cognitive bias “positive illusion,” a systemic decision/judgment error of “unrealistic optimism about the future,” that we addressed in our November 11, 2020 article about medtech and cognitive biases.
Hoping to help medtech companies leveraging advanced computing technologies better mitigate their cyber risks, this article has three parts:
- It details the complex, business-threatening cyber risks that digital medtech companies face;
- It discusses the four factors exacerbating these already business-threatening risks; and
- It provides a chilling, realistic cyber-attack scenario that brings it all together.
Medtech’s Complex, Business-Threatening Risks
To help medtech companies understand their business-threatening cyber risks, we offer an infographic and related taxonomy classifying cyber risks into five categories of harmful events — operational interference, device problems, data breaches, financial and IP theft, and compliance and sales issues — and showing that one or more harmful events can produce one or more types of financial harm: lost sales, customers, investors, and partners; regulatory costs; extortion payments; financial theft; and investigation and remediation costs.
You don’t need a working knowledge of the cyber-attacks in the top gray box of the figure above. But you should know that the following:
- Cyber-attacks can cause one or more harmful events.
- Harmful events can inflict one or more types of financial harm.
- Cyber criminals are quickly and continuously finding new vulnerabilities (as this NIST graph on CVSS Severity over time shows), developing new cyber-attacks, and crafting new ways to use cyber-attacks to make money.
- To cost-effectively mitigate these risks, you need a multi-tool, multidisciplinary cyber risk mitigation system — as our third article in this series will explain.
The following summarizes the harmful events that medtech companies can suffer from cyber-attack, and the related financial harms.
Harmful Event Categories
1. Operational Interruptions: An example is the June 1, 2020, ransomware attack on the University of California San Francisco that encrypted data in its School of Medicine for which the institution paid a $1.14 million ransom. The school also incurred significant investigation costs and untold harm from reputational damage. A reputable cybersecurity company, Sophos, reported that “ransomware attacks on healthcare almost doubled in 2021 (66% in 2021 versus 34% in 2020)” and that “the average remediation cost went up from US$1.27M in 2020 to US$1.85M in 2021.” However, these costs underrepresent represent the long-term damage from reputational harm that can be business-threatening to digital medtech companies. The year 2022 might be even worse, because from January to June, HHS’s Office of Civil Rights, the agency’s investigatory and enforcement arm, reported “256 hacks and information breaches, up from 149 for the same period a year ago.” And the most expensive attack against a medtech/pharma company was the 2017 NotPetya debacle at Merck, for which the company publicly reported that the attack inflicted about $1.3 billion in damage.
2. Device Problems: As we use the term, device problems come from both cyber-incidents that exploit cyber vulnerabilities and just from the existence of those vulnerabilities. Just the existence of vulnerabilities can prevent premarket device approval, interfere with postmarket sales, and/or inflict significant postmarket costs from remediating device vulnerabilities, reputational harm from notifying the public about vulnerabilities, and business-threatening recalls. All these can come with huge costs.
Medtech companies face even greater financial risks when cyber-incidents exploit device vulnerabilities that lead to patient harm. In these instances, the liability is virtually unlimited. According to the e-book “Healthcare Security Woes Balloon in a Covid-Era World,” in 2020, the U.S. Cybersecurity and Infrastructure Security Agency (CISA) issued more than a half-dozen warnings for infusion pumps and the FDA issued a series of warnings advising medical device manufacturers to secure their devices against various vulnerabilities, including Log4Shell, SweynTooth, URGENT/11, Ripple20, and SigRed. The Ripple20 vulnerabilities are an example of a common, scary, and potentially devastating medical device vulnerability. With new technologies and new devices come new means by which vulnerabilities can be exploited. Consider this NIST chart showing the rapid growth in computing vulnerabilities from 2001 to 2022. (For more information about medtech computing vulnerabilities, try this helpful white paper from Medcrypt.) Moreover, there is a robust plaintiffs’ bar with the resources and expertise ready to bring class actions and individual suits against medtech companies whose devices harm people.
3. Data Breaches: Examples include the 218 million accounts stolen from Zynga in 2019, the 538 million accounts stolen from Sina Weibo (the Chinese Twitter) in 2020, the 162 million user accounts stolen from Dubsmash in 2018, and the 150 million user accounts stolen from My Fitness Pal in 2018. Even one of the ultimate tech companies, Microsoft — with almost unlimited security resources — suffered a data breach of 250 million customer records in 2019. Medtech and healthcare providers suffer data breaches at alarming rates. Consider perusing the many breach investigations by HHS’s Office for Civil Rights, which shows 650 data breaches affecting 500+ individuals in 2020 and 714 in 2022. Data breaches cost companies millions. The Ponemon Institute’s well-respected report stated that the average United States data breach in 2020 cost $8.64 million. Additionally, smaller breaches like the 1.12 million records stolen from Washington State University in 2019 inflict business-killing damage. Washington State had to pay $4.7 million to settle a class action brought by the data breach victims, and in 2015, the primary teaching hospital for the UCLA school of medicine settled its data breach lawsuit for $7.5 million.
4. IP & Financial Theft: Digital medtech companies are prime targets for intellectual property theft. This includes the IP theft by foreign government-sponsored attackers, particularly the Chinese, that the U.S Intelligence Director warned about in December 2020. He said that the “intelligence is clear: Beijing intends to dominate the U.S. and the rest of the planet economically, militarily and technologically.” He described China’s approach of economic espionage as “rob, replicate, and replace,” saying that “China robs U.S. companies of their intellectual property, replicates the technology, and then replaces the U.S. firms in the global marketplace.” Moreover, this IP theft is the primary goal of Hafnium, the cyber-attack group in China responsible for the Microsoft Exchange Server hacks, as discussed below.
5. Sales, Partners, & Compliance Issues: Two of the biggest cyber risks for medical device companies are problems and delays in device approval related to premarket cybersecurity requirements and inability to prove your device’s cybersecurity to customers and partners — who often apply more stringent cybersecurity requirements than the regulators. Digital medtech companies need to understand the market demand for their security, including how they will answer the questions in the Manufacturer Disclosure Statement for Medical Device Security (MDS2), which many large healthcare providers will require before buying your devices. Plus, when setting their cyber risk mitigation strategies, medtech companies need to consider the true potential cost of their cyber risks — which covers all premarket and postmarket costs — which this article helps them do. Another form of partner cyber risk arises through your supply chain: e.g., software or hardware that you incorporate in your devices have computing vulnerabilities that require expensive remediation and/or lead to device malfunctions that injure patients.
Financial Harm Types
1. Lost Sales, Customers, Investors, & Partners: These comprise the costs when harmful event(s) lead to the loss of sales, customers, investors, and partners. In many situations, medtech companies will be required to notify people and/or regulators about the harmful events:
- HIPAA Covered Data Breaches: Many medtech companies are covered by the HIPAA, and the HIPAA Breach Notification Rule, 45 CFR 164.400-414, requires notification to HHS and the public following a breach of unsecured protected health information.
- Data Breaches for Public Companies: The SEC requires public companies to provide notice about any cyber-incident, including data breaches, that meet the materiality standard. In March 2022, the SEC proposed even more stringent data breach reporting requirements.
- FDA Required Reporting: The Medical Device Reporting (MDR) regulation (21 CFR Part 803) requires device manufacturers to report to the FDA when they learn that any of their devices may have caused or contributed to a death or serious injury. The FDA’s Postmarket Management of Cybersecurity in Medical Device guidance requires that device manufacturers report uncontrolled residual risks relating to cyber-incidents and/or cyber vulnerabilities.
- Non-Healthcare Privacy Laws: The GDRP and every state has a privacy law that might require notification.
Therefore, after almost every material cyber-incident your company experiences, your prospects, customers, investors, and partners will find out what happened. In many situations, learning about your cyber-incident will degrade others’ confidence in you in ways that cost you sales, customers, investors, and partners. Much worse are device problems that injure patients, as these harmful events can decimate your reputation and produce staggering liability via class actions or individual lawsuits.
2. Investigation & Remediation Costs: These comprise your costs to investigate the cyber-attack that caused the harmful event(s) and to remediate your operations and/or offerings to at least their original form. These costs can be quite high. As noted above, Sophos found that “the average remediation cost [for healthcare companies hit by ransomware] went up from US$1.27M in 2020 to US$1.85M in 2021.” An additional major financial risk is that some computing vulnerabilities — even if never actually exploited by a cyber-incident — will be so egregious that the FDA will require expensive remediation and/or recalls.
3. Legal & Contract Costs: These comprise your costs from harmful events breaching existing contracts (e.g., SLAs) and/or leading to lawsuits against you. Many medtech companies fail to consider these costs when setting their cyber risk strategies. To avoid this, as article 3 will explain, medtech companies need to deploy a multidisciplinary approach to cyber risk mitigation.
4. Regulatory Costs: These comprise the costs when any relevant regulatory entity investigates your cyber-incident, takes negative actions against you (e.g., fines or enforcement action), and/or requires your company to remediate the harm from a cyber-incident. There’s a slight overlap with investigation and remediation costs, though companies that have cyber-incidents will almost always have investigation and remediation costs but not necessarily regulatory costs.
5. Extortion Payments: These are the costs of paying ransom to cyber-attackers for the means to restore your operations and/or prevent the distribution of information stolen from you.
6. Financial Theft: These are the direct monetary losses when an attacker steals money from you.
4 Factors Exacerbating Cyber Risks
Unfortunately, as this infographic below introduces, four factors are significantly exacerbating medtech’s cyber risks:
Increasingly Rigorous Regulations: Regulators in the United States, the EU, and around the world are continuously making their medical device cybersecurity requirements more rigorous. The prime examples are EU’s regulations on medical devices 745/2017 (MDR) and on in vitro diagnostic medical devices 746/2017 (IVDR), both adopted and entered into force on May 25, 2017. The MDR took full effect on May 26, 2021, and the IVDR on May 26, 2022. A good explanation of these more rigorous medical device requirements can be found in the MDCG 2019-16 Guidance on Cybersecurity for medical devices.
In April 2022, the FDA published a draft “premarket cybersecurity” guidance that supersedes its 2018 draft premarket cybersecurity guidance. The guidance covers all devices that contain software, firmware, programmable logic, and Software as a Medical Device (SaMD). Highlights of this more rigorous regulatory scheme include:
- Increased emphasis on how cybersecurity relates to the FDA’s Quality System Regulation (QSR), including recommending the use of Secure Product Development Frameworks (SPDF) to achieve that goal. The SPDF comprises all aspects of a device’s total product life cycle, including development, release, support, and decommission.
- Recommendations that the design process include threat modeling be performed in the design process: e.g., how cyber-attackers could attack the device and how the manufacturer would prevent such attacks.
- Recommends a Software Bill of Materials (SBOM) instead of a Cybersecurity Bill of Materials (CBOM). FDA states that the SBOM should include software component descriptors (name, version, manufacturer), level of support provided, end of support date, and any known vulnerabilities.
- While it removed the requirement for manufacturers to categorize their product into risk tiers, it requires them to provide much more detail in their premarket submissions, including providing technical manuals that healthcare providers can use to efficiently patch device vulnerabilities.
While the new draft guidance is voluntary, the FDA does state, “not following the guidance will create greater, probable complexities or potential hardships as far as addressing questions that will come up during the review process. That means potential delays.” This makes it essentially and practically “involuntarily voluntary.”
Large, Growing Attack Surface & Computing Tech Dependency: By their nature, medtech companies strive to innovate and reduce costs by continuously adding more computing technologies, particularly in these areas:
- Increased computing power/internet access and new software applications and hardware to deliver their innovative offerings.
- The dissolution of the traditional security perimeter through cloud storage, mobile devices, and medtech systems in patients’ homes.
- Sophisticated data analytic technologies (e.g., machine learning and AI) needed to disrupt markets and overcome incumbents.
Each addition of computing technology heightens a company’s cyber-attack risks by increasing both its cyber-attack surface (i.e., the sum of physical and digital vulnerabilities that can be exploited to carry out a cyber-attack against you) and its dependency on computing technologies. Unfortunately, the attack surface and dependence on computing technologies of today’s digital medical device companies is enormous and growing at a dangerous time: namely, the looming, major cybercrime way explained below. This stark reality, in addition to other factors, strongly favors incorporating multidisciplinary cyber risk mitigation strategy into your product development process and use of any computing technologies, as article 3 will explain.
High Cybersecurity Professional Churn: The average tenure of chief information security officers (CISOs) in all industries is 26 months, according to the Nominet CISO Stress Report. This churn is a serious problem, both creating high talent search fees and leaving the companies less protected compared with reliable, long-term cybersecurity leadership. Consider these “Cybersecurity Workforce Demand” facts and the 2021 Cybersecurity Workforce Study.
Major, Looming Cybercrime Wave: The economic incentives for cyber criminals are the highest in history, are growing, and are about to generate an unprecedented wave of extortion-based cybercrime, often referred to as ransomware. Most digital medical device leaders understand ransomware basics – namely, that upon infection, it uses encryption to incapacitate computers followed by a payment demand for the means to restore operations, or even destructive action to render a device useless (e.g., NotPetya). But few leaders know that the recent trend of paying ransoms has created a vibrant ransomware ecosystem that even includes the sale of ransomware software-as-service platforms that come with free trials and tech support. Combined with the rapid increase in the use of computing technologies around the world, this vibrant ecosystem is about to generate an unprecedented wave of extortion-based cybercrimes exactly when academic institutions have scant money for security/prevention and keep losing cybersecurity personnel at the highest rate ever.
Moreover, cyber-attacks are becoming ever-more sophisticated, such as the recent Solarwinds debacle and Microsoft Exchange Server hacks. In early 2020, attackers hacked Solarwinds’ software development environment and installed a sophisticated cyber-attack tool (called a backdoor) in the update to Solarwinds’ widely used IT monitoring software. By updating their Solarwinds software, customers installed a backdoor that allowed the attackers to install additional malware and launch more attacks. Because the overall Solarwinds attack was so sophisticated it went undetected for months; compromised the computing systems of at least 18,000 Solarwinds customers, including Fortune 500 companies, many U.S. government agencies, and some universities and colleges, and no one knows the extent of the damage.
In early January 2021, cybersecurity experts learned that cybercriminals were actively exploiting four newly discovered vulnerabilities in multiple versions of Microsoft Exchange Server — email and calendaring software widely used around the world. These vulnerabilities helped the attackers steal information and install additional attack tools that facilitate long-term access to the victim’s computing networks. According to Microsoft VP Tom Burt, the main cybercriminal group exploiting the vulnerabilities, known as Hafnium, is a “highly skilled and sophisticated” “state-sponsored” group operating from China that “primarily targets entities in the United States” to steal “information from a number of industry sectors, including infectious disease researchers, law firms, higher education institutions, defense contractors, policy think tanks, and NGOs.” While Microsoft on March 2, 2021 released “security updates” to address these vulnerabilities, the attackers had a long time to use the vulnerabilities to steal valuable information and install covert attack tools. Moreover, according to RiskIQ, 82,731 instances of this software remained insecure (i.e., not updated) as of March 11, 2021. Finally, Microsoft believes that it took about 1,000 software engineers just to create the Solarwinds attack, a massive criminal effort.
The Solarwinds debacle and the Microsoft Exchange Server hacks, therefore, emphasize the gravity of the cyber risks for medtech.
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

17 Sep
The 5 Common Medical Device Startup Challenges (& How To Overcome Them)
Peggy Fasano, chief operating officer at Boulder iQ, wrote in the current issue of meddeviceonline: “The medical device market is diverse, fast-moving, and powered by dynamic customer needs and ongoing demand. Anticipated to reach $734.39 billion by 2027, the market is a land of both opportunity and risk for enthusiastic, innovative developers. Understanding the most common challenges device developers face, and how to successfully navigate them, can provide the competitive edge an entrepreneur needs to succeed in this environment.
1. Understanding And Accepting That Time Is Money
In medical device development, the old adage, “time is money,” is a constant. The sooner you can get a device on the market, the sooner it moves from an expense to a source of revenue. The quicker you get to market, the less risk you have of a competitive product or technology usurping yours. Many companies today also find that the longer their time to market stretches, the more problems they have in acquiring the parts and materials they originally specified in design.
Speeding time to market also can provide more opportunity for partnerships with other companies and for potentially integrating your technology into other devices. In short, there’s a lot to be said for being the market leader in a given segment.
How To Overcome The Challenge
The first step in meeting the challenge is to acknowledge that getting a device from prototype to market is going to take more time than you think. The second step is to truly understand each step of the process and accept that you will be unable to handle everything on your own.
In order to determine the path and timeline your device will take, you must be very clear – up front – on the problem you are looking to solve, on your potential customer base, and on the market for the device aiming to solve that problem. Get out and talk to potential users to get crystal clear on what they need. Putting in that effort will help ensure that the time and money you spend in developing your device will produce maximal return.
Determine needed assumptions and analyze the risks. Then decide which steps you can perform in parallel and which you’ll need to do sequentially. Pay particular attention to those with the longest lead times. Accept that 0% risk is unrealistic for any medical device and that you will be unable to perform every step on your own. Know when to bring in help.
2. Acquiring The Needed Breadth Of Resources
It is no secret that the breadth of resources required to get a medical device to market is daunting. Expertise in project management, product design and engineering, regulatory strategies and submissions, quality managements systems and quality assurance support, manufacturing services, sterilization, and packaging is critical. In fact, one of the FDA’s expectations is that medical device personnel are qualified and competent to perform their assigned functions – no small feat in a startup or small business.
The real challenge is in how best to acquire this expertise. Many device developers attempt to handle many of the functions on their own. Particularly with smaller businesses, owners understandably wear many hats. But whether the business is large or small, the reality is that no one person can wear them all. And, the challenge in acquiring talent has become bigger in today’s tight labor market, where it’s difficult to source, hire, train, and retain qualified staff.
How To Overcome The Challenge
Because time to market is critical, finding the specific services and personnel to expedite your commercialization efforts must be a top priority.
Assess if it makes sense for you to bring your product development needs in-house or if contracting subject matter experts in designated areas will make better use of your limited dollars. If you can support bringing some services in-house, make sure those individuals will be able to flexibly interact with contracted subject matter experts in other areas. Also consider bringing in quality and regulatory training necessary to lay a solid foundation from which your company can grow.
If you go the contract route, find a service provider with the experience to streamline your product development efforts from prototype through commercialization – and beyond.
In short, don’t spend a dime until it is time, but when it is time, reach out to those who have the experience to assist.
3. Evaluating And Managing Vendors For Competence & Efficiency
It is not unusual for a device developer to engage with a contract manufacturer or to work with a variety of subject matter experts and sources for different parts of the development process. Evaluating, qualifying, working with, and managing multiple vendors can be draining – of both time and money necessary to support long-term company objectives.
Device developers also face a challenge in making sure that all vendors are aligned and similarly motivated on the singular common goal of getting their device to the market as quickly and efficiently as possible. Today, they face the added challenge of making careful choices to avoid supply chain issues to the extent possible. Finding contracted vendors who communicate and coordinate well with you, and with other vendors, is crucial in getting a device to market and in achieving standardization of processes and procedures so that the device meets its intended use every time.
How To Overcome The Challenge
A single-source expert contract consulting firm may be the most cost- and time-efficient way to handle both this challenge and the challenge of finding needed breadth and depth of expertise. Working with established systems, processes, and relationships, a single-source contract firm will provide a team of experts who know the ins and outs of bringing a device to market.
While expert contract consulting firms sometimes work with contractors themselves, it can be helpful to find a firm with a stable of in-house expertise. This type of team will be able to step in and work together seamlessly. They often can bring on board a proven ISO 13485-certified quality management system, too, that allows simple and standardized records, documentation, and training management for the entire development process.
If you go this route, thoroughly vet any potential firm for experience and a track record of success. Make sure you will be dealing with the actual experts who have worked in and with device firms. If they’ve walked in your shoes, they will be best-positioned to help you avoid missteps and get you on the clearest, straightest, most time-effective path to market.
4. Tackling Regulatory Hurdles
Dealing with regulatory requirements requires specific knowledge, expertise, and experience in working deeply with device development firms and with regulatory agencies. Why? Because you can design, manufacture, and validate your medical device – and even secure intellectual property protections – and still face the reality that you also must determine the most efficient pathway to regulatory clearance and/or authorization.
Key to regulatory submission efficiencies and cost savings is understanding how to determine product codes, when to employ the 513(g) process, and how to use the FDA Q-Sub program to speed time to market. Add in more complicated needs, such as clinical trial management or international medical device registration and licensing, and the risks of a mistake loom large.
How To Overcome The Challenge
Outsourcing the regulatory affairs function is typically the most effective way for startup and smaller companies to obtain the required experience and expertise. Here, again, it is critical to work with people who have “been there, done that” more than once. Whether it’s determining classification of the product, communicating with the FDA, or making calls on expensive testing, experience really does matter.
Beyond bringing pure legal or regulatory knowledge to the table, experienced regulatory affairs experts will know how, when, and where to integrate regulatory affairs throughout the development process. What may seem obvious in regulatory approval often isn’t. For example, experienced professionals might determine that a seemingly straightforward regulatory pathway will actually work better as a series of multiple smaller steps.
Or, they may know that tweaking a device’s intended use and/or indications for use or removing a function or feature could be valuable in getting the device through the regulatory process and into the market faster. The resulting initial device may appeal to early adopters and provide all the benefits of market introduction – including revenue. Later, with a footing in the market and an established safety profile, you can go back and add features and functions through abbreviated pathways.
Only a professional – or team of professionals – with solid experience in device development will be able to identify and implement regulatory paths in this way, with focus on speeding products to market and obtaining the greatest return on investment.
5. Managing The Budget
No matter what the device, how brilliant the inventor is, or how sharp that individual’s management skills, it takes significant budget to get through the entire development process for any product. There is no shortcut to quality and success, and the cost of poor planning, repeat testing, and/or noncompliance can quickly erode even the most carefully planned budget.
How To Overcome The Challenge
The tendency of device firms is for the founder and/or device inventor to handle the finances. If this is the case, the founder needs to carve out time to really think through the budget and be wholly responsible for it. The founder needs to ensure funds are being allocated appropriately between product development, marketing, sales, and other areas.
Too often, funds are not carefully controlled, and budget is used in areas where it is not necessary or not yet necessary. An example of this is building up a sales team too early. If something happens to the schedule and the market release is delayed, you now have an entire sales team that is on the payroll. Another example is purchasing manufacturing equipment that will be needed for production while still in the design phase. Do you really need that automated equipment for a pilot build of 50 units? Think hard in terms of “need” or “want” when it comes to any purchase or use of funds.
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

23 Jun
FDA Releases Guidance On Cybersecurity In Medical Devices
John Giantsidis, president, CyberActa, Inc., wrote in the current issue of meddeviceonline: “The digital revolution that resulted in the Internet of Things (IoT), Internet of Medical Things (IoMT), Software as a Medical Device (SaMD), and connected devices permeating the healthcare environment, both in the hospital and at home, comes with the possibility of cyberattacks and intrusions against a compromised connected medical device and the network to which such a device is connected.
Healthcare has been proven to be a valuable target for cyber threat actors (also known as hackers) and medical devices are increasingly the targets of malicious cyberattacks, which result not only in data breaches but also increased healthcare delivery costs, and they can ultimately affect patient health outcomes.
The consequences of these attacks, and the corresponding safety and fiscal impacts that can result from them, have prompted many governments agencies and other actors (industry associations, technical societies, standards organizations, research institutions, policy groups, and nongovernmental organizations) to undertake measures to protect themselves and their citizens. In the EU, both the MDR and IVDR requirements mandate consideration of medical device cybersecurity, and the Medical Device Coordination Group (MDCG) in its guidance directs manufacturers on how to fulfil all the relevant essential requirements of Annex I to the MDR and IVDR with regard to cybersecurity.2 In Australia, the Therapeutics Goods Administration is treating medical device cybersecurity as part of the Essential Principles and the TGA requires that the cybersecurity “Essential Principles are met by applying accepted best-practice regarding quality management systems and risk management frameworks, which is typically via application of state of the art standards.”
Since 2005, the FDA has been striving to enhance medical device cybersecurity, and the latest FDA effort is a draft guidance that expects security throughout the total product life cycle (TPLC). Another effort is bipartisan congressional support of the Protecting and Transforming Cyber Health Care Act of 2022 (PATCH Act of 2022),4 which, if passed, would revise the existing Federal Food, Drug, and Cosmetic Act. However, let’s revisit the PATCH Act if/when it gets passed.
The FDA’s draft guidance on medical device cybersecurity provides insight on how the agency is going to apply the existing regulatory requirements. But how does one get ready to meet FDA’s medical device cybersecurity expectations?
First, it is important to understand that the scope of FDA’s guidance is exceptionally broad and covers devices that contain software (including firmware) or programmable logic, as well as SaaMD, and would be expected for:
- Premarket Notification (510(k)) submissions
- De Novo requests
- Premarket Approval Applications (PMAs) and PMA supplements
- Product Development Protocols (PDPs)
- Investigational Device Exemption (IDE)/Humanitarian Device Exemption (HDE) submissions
- All devices within the meaning of the Federal Food, Drug, and Cosmetic Act (FD&C Act), whether or not they require a premarket submission.
Principles Of Medical Device Cybersecurity
The FDA guidance establishes six broad expectations and introduces the newly minted concept of a Secure Product Development Framework (SPDF), which encompasses all aspects of a product’s life cycle, including development, release, support, and decommission to satisfy Quality System Regulations (QSR) in 21 CFR Part 820:
- Cybersecurity is an integral part of device safety and the QSR
- Security by design
- Transparency
- Security risk management
- Security architecture
- Testing/objective evidence
Cybersecurity Is An Integral Part Of Device Safety And The QSR
The FDA guidance sets the nexus between a reasonable assurance of safety and effectiveness for devices with cybersecurity risks and the expectation that such cybersecurity risks are governed by the QSR. The FDA borrows from NIST and introduces a Secure Product Development Framework (SPDF) concept, akin to NIST’s Secure Software Development Framework5 and suggests that such integration with product and software development, risk management, and the quality system at large can satisfy cybersecurity QSR. The following principles may be considered the fundamental practices that the FDA had in mind regarding SPDF:
- Define Security Requirements for Product Development: Ensure that security requirements for product development are known at all times so they can be taken into account throughout TPLC and duplication of effort can be minimized because the requirements information can be collected once and shared. This includes requirements from internal sources (e.g., the organization’s policies, business objectives, and risk management strategy) and external sources (e.g., applicable laws and regulations).
- Implement Roles and Responsibilities: Ensure that everyone inside and outside of the organization involved in the TPLC is prepared to perform their TPLC-related roles and responsibilities throughout the TPLC.
- Implement Supporting Toolchains: Use automation to reduce human effort and improve the accuracy, reproducibility, usability, and comprehensiveness of security practices throughout the TPLC, as well as provide a way to document and demonstrate the use of these practices.
- Define and Use Criteria for Product Security Checks: Help ensure that the product resulting from the TPLC meets the organization’s expectations by defining and using criteria for checking the product’s security during development.
- Implement and Maintain Secure Environments for Product Development: Ensure that all components of the environments for product development are strongly protected from internal and external threats to prevent compromises of the environments or the product being developed or maintained within them.
- Protect All Forms of Code from Unauthorized Access and Tampering: Help prevent unauthorized changes to code, both inadvertent and intentional, which could circumvent or negate the intended security characteristics of the product.
- Provide a Mechanism for Verifying Product Release Integrity: Help product/software acquirers ensure that the product/software they acquire is legitimate and has not been tampered with.
- Archive and Protect Each Product Release: Preserve product/software releases in order to help identify, analyze, and eliminate vulnerabilities discovered in the product/software after release.
- Design Product to Meet Security Requirements and Mitigate Security Risks: Identify and evaluate the security requirements for the product; determine what security risks the software is likely to face during operation and how the software’s design and architecture should mitigate those risks; and justify any cases where risk-based analysis indicates that security requirements should be relaxed or waived.
- Review the Product Design to Verify Compliance with Security Requirements and Risk Information: Help ensure that the product will meet the security requirements and satisfactorily address the identified risk information.
- Reuse Existing, Well-Secured Software When Feasible Instead of Duplicating Functionality: Lower the costs of software development, expedite software development, and decrease the likelihood of introducing additional security vulnerabilities into the product by reusing software modules and services that have already had their security posture checked. This is particularly important for software that implements security functionality, such as cryptographic modules and protocols.
- Create Source Code by Adhering to Secure Coding Practices: Decrease the number of security vulnerabilities in the software and reduce costs by minimizing vulnerabilities introduced during source code creation that meet or exceed organization-defined vulnerability severity criteria.
- Configure the Compilation, Interpreter, and Build Processes to Improve Executable Security: Decrease the number of security vulnerabilities in the software and reduce costs by eliminating vulnerabilities before testing occurs.
- Review and/or Analyze Human-Readable Code to Identify Vulnerabilities and Verify Compliance with Security Requirements: Help identify vulnerabilities so that they can be corrected before the software is released to prevent exploitation.
- Test Executable Code to Identify Vulnerabilities and Verify Compliance with Security Requirements: Help identify vulnerabilities so that they can be corrected before the product is released in order to prevent exploitation.
- Configure Product to Have Secure Settings by Default: Help improve the security of the product at the time of installation to reduce the likelihood of the product being deployed with weak security settings, putting it at greater risk of compromise.
- Identify and Confirm Vulnerabilities on an Ongoing Basis: Help ensure that vulnerabilities are identified quickly so they can be remediated quickly in accordance with risk, reducing the window of opportunity for attackers.
- Assess, Prioritize, and Remediate Vulnerabilities: Help ensure that vulnerabilities are remediated in accordance with risk to reduce the window of opportunity for attackers.
- Analyze Vulnerabilities to Identify Their Root Causes: Help reduce the frequency of vulnerabilities in the future.
Security By Design
The FDA guidance sets the expectation that devices must meet the security objectives of authenticity, integrity, authorization, availability, confidentiality, and secure and timely updatability and patchability. The extent to which security requirements are needed to meet these objectives would depend on:
- the device’s intended use and indications for use,
- the presence and functionality of its electronic data interfaces,
- the intended and actual environment of use,
- the type of cybersecurity vulnerabilities present,
- the exploitability of the vulnerabilities, and
- the risk of patient harm due to vulnerability exploitation.
For example, a device would be deemed that it appropriately accounts for authenticity when it evaluates and ensures authenticity for:
- information at rest (stored),
- information in transit (transmitted),
- entity authentication of communication endpoints, whether those endpoints consist of software or hardware,
- software binaries,
- integrity of the execution state of currently running software, and
- any other appropriate parts of the system where a manufacturer’s threat model and/or risk analyses reveal the need for it.
The expectations for demonstrating Integrity (Code, Data or Execution) provide an illustrative example:
Code
Verify authentication tags (e.g., signatures, message authentication codes [MACs]) of software/firmware content, version numbers, and other metadata.
Install cryptographically authenticated firmware and software updates and do not allow installation where such cryptographic authentication either is absent or fails. Use cryptographically signed updates to help prevent any unauthorized reductions in the level of protection by ensuring that the new update represents an authorized version change.
Ensure that the authenticity of software, firmware, and configuration are validated prior to execution, e.g., “allow-listing” based on digital signatures.
Disable or otherwise restrict unauthorized access to all test and debug ports (e.g., JTAG, UART) prior to delivering products.
Employ tamper evident seals on device enclosures and their sensitive communication ports to help verify physical integrity.
Data
Verify the integrity of all incoming data, ensuring that it is not modified in transit or at rest.
Validate that all data originating from external sources is well-formed and compliant with the expected protocol or specification. Additionally, validate data ranges to ensure they fall within safe limits.
Protect the integrity of data necessary to ensure the safety and effectiveness of the device.
Execution
Verify integrity of code while it is being executed on the device.
Review all code that handles the parsing of external data using automated (e.g., static and dynamic analyses) and manual (i.e., code review) methods.
Transparency
The FDA guidance concludes that transparency is critical to ensure safe and effective use and integration of devices and systems and such transparency can be conveyed via both device labels and vulnerability management plans.
For devices with cybersecurity risks, FDA also believes that informing users of security information through labeling may be an important part of QSR design controls to help mitigate cybersecurity risks and help ensure the continued safety and effectiveness of the device. A word of caution, however: Any risks transferred to the user should be detailed and considered for inclusion as tasks during usability testing (e.g., human factors testing) to ensure that the user has the capability to take appropriate actions to manage those risks. The FDA outlines the following labeling to communicate security information to users:
- Device instructions and product specifications related to recommended cybersecurity controls appropriate for the intended use environment (e.g., anti-malware software, use of a firewall, password requirements).
- Sufficiently detailed diagrams for users that allow recommended cybersecurity controls to be implemented.
- A list of network ports and other interfaces that are expected to receive and/or send data.
- User guidance regarding supporting infrastructure requirements so that the device can operate as intended (e.g., minimum networking requirements, supported encryption interfaces).
- A Software Bill of Materials (SBOM) in a format for users to effectively manage their assets, to understand the potential impact of identified vulnerabilities to the device (and the connected system), and to deploy countermeasures to maintain the device’s safety and effectiveness.
- A description of systematic procedures for users to download version-identifiable manufacturer-authorized software and firmware, including a description of how users will know when software is available.
- A description of how the design enables the device to respond when anomalous conditions are detected (i.e., security events) to maintain safety and effectiveness.
- A high-level description of the device features that protect critical functionality (e.g., backup mode, disabling ports/communications, etc.).
- A description of backup and restore features and procedures to restore authenticated configurations.
- A description of the methods for retention and recovery of device configuration by an authenticated authorized user.
- A description of the secure configuration of shipped devices, a discussion of the risk trade-offs that might have been made about hardening options implemented by the device manufacturer, and instructions for user-configurable changes.
- A description of how forensic evidence is captured, including but not limited to any log files kept for a security event. Log file descriptions should include how and where the log file is located, stored, recycled, archived, and how it could be consumed by automated analysis software.
- Technical instructions to permit secure network deployment and servicing, and instructions for users on how to respond upon detection of a cybersecurity vulnerability or incident.
- Information, if known or anticipated, concerning device cybersecurity end of support and end of life. If the device remains in service following the end of support, the manufacturer should have a preestablished and pre-communicated process for transferring the risks highlighting that the cybersecurity risks for end users can be expected to increase over time.
- Information on securely decommissioning devices by sanitizing the product of sensitive, confidential, and proprietary data and software.
The FDA guidance further expects that manufacturers institute a formal plan for how they will identify and communicate vulnerabilities that are identified after releasing the device to users. This plan is to be in unison with risk management processes under 21 CFR 820.30(g) and corrective and preventive action processes in accordance with 21 CFR 820.100.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

09 Jun
Key Design Considerations For Digitally Connected Injectors
Jason Song, P.E., co-founder and chief technology officer of SureMed Technologies, Inc., wrote in the current issue of meddeviceonline: “Digital health has become an integral part of clinical trials and the commercial patient support ecosystem. In the past two years, there has been a significant transformation in the way clinical trials are conducted, as well as how healthcare providers support patients and patients’ expectations around their illness management. With COVID-19 and the public health lockdowns and social distancing measures, clinical trials leaped from traditional in-person interactions between investigators and trial participants to a more virtual setting with an open embrace of digital health and telemedicine tools. With social distancing and challenges around in-person visits to clinics, insurers, healthcare providers, and patients have also embraced and championed the use of digital and virtual tools. Today, doctors are more accepting of reviewing a patient’s heart monitor data generated by the patient’s Apple smartwatch. In fact, talking to healthcare providers, doctors, and nurse practitioners reveals that digital tools provided more value and unexpected benefits, such as the ability to see a broader view of patients’ health situations over time between visits.
Pharmaceutical companies have also seen the benefit of integrating digital capabilities within clinical trials. First, no longer is recruitment hindered by geolocation and proximity to trial sites. In addition, for the past five or so years, pharmaceutical companies have seen the benefit of integrating digital tools and technologies as part of a digital patient support ecosystem. The tools and technologies allow for better patient engagement and treatment support that translate to stronger brand position, real-world evidence, and deeper patient insights. In the case of inhaled products such as Symbicort, digital connectivity through inhaler add-ons or integration has demonstrated improvement in patient disease management through improved adherence and compliance. In addition, data from connected inhalers, such as the Turbohaler from AstraZeneca, have generated real-world information, such as the number of user days on treatment, as well as helping to identify predictors of asthma attacks and/or disease exacerbations.
Today, we see wide adoption of digital inhalers, and many newly launched inhaled products have digital connectivity capabilities that integrate them into a patient support digital ecosystem. This, unfortunately, is not the case in the injectable drug delivery device space. Today’s injection drugs are still being developed and launched without a true connected device element to track adherence and compliance. Worse yet, while many inhaled products are going into clinical trials with connected inhalers to help with trial adherence and compliance as well as support clinical trial endpoints, there are no connected injectors going into clinical trials. As connected drug delivery devices require clinical trial data to support their use and benefit before they can be submitted for regulatory approval, this means we most likely will not see connected injection devices, such as prefilled syringes, autoinjectors, or needle safety devices that are widely used for biological drug products, for some time. The exception is in the diabetes space, where we have seen a wider advancement in pen or multi-dose injector digital connectivity, as well as one or two products that utilize expensive syringe injection aid/assistance devices, such as the syringe injection assistance device for Betaseron, whereby the user loads the syringe onto a battery and motor operated injection assistance device to help with injection.
With the wide success of connected inhalers, it raises the question: Why aren’t there any successful connected injectable devices despite the many companies offering connectivity accessories for injector devices? To answer this question, we will have to dive into what makes a successful connected drug delivery device.
Make Sure Your Connected Injector Fits Within A Digital Ecosystem
A successful connected drug delivery device must first and foremost fit within a digital ecosystem, whether it is a clinical trial ecosystem or a commercial patient support ecosystem. The connected drug delivery device is a special case, as its role within both the clinical and commercial digital ecosystem is to confirm that the patient has taken their medication. As such, it requires the same connected device to be used in the commercial environment as was used to demonstrate adherence and compliance in clinical trials. In the case of a connected drug delivery device, any device data captured beyond whether the medication was administered or not provides no additional added value and will oftentimes throw the digital ecosystem out of balance, resulting in a detrimental hindering effect. A digital ecosystem can be viewed like a watch; each component must serve its purpose exactly, no more and no less. When one piece over- or underperforms, it will throw the system out of balance, just as an incorrect gear in a watch will render the watch inaccurate and, over time, nonfunctional and abandoned.
Similarly, additional information or data gathered by a digital device beyond its purpose can create unnecessary confusion and problem for the patient, user, and/or parties involved with treatment management. Using another analogy, the speedometer on a car serves only one purpose and that is to tell you how fast you are driving. It needs to function exactly as intended for the driver. Capturing too much information, such as how bumpy the road is, how straight you are driving, the frequency of acceleration and brake usage, etc., will not help but will only distract the driver from the critical speed information that the driver needs to maintain safe driving. This is why, at the moment, cars have hands-free call features but no features to support texting or IMs.
To this end, I will start off by first analyzing why connected inhalers and connected insulin pen injectors, by and large, have been successful, while connected syringe and autoinjector connectivity solutions by large have missed the mark and have largely been rejected and eventually abandoned by pharmaceutical manufacturers.
Connected inhalers and connected insulin pen injector add-ons and solutions have been successful because those solutions provide the balanced data to fit within their respective digital ecosystems. And in doing so, they demonstrate their value in helping patients stay adherent to their medication regimen. Connected insulin pen injectors have been proven to help patients maintain and keep their H1Ac levels in check and, therefore, help slow disease progression and minimize health complications that are associated with uncontrolled diabetes. These devices, combined with artificial pancreas systems, have revolutionized diabetic care management and patient enablement. The same goes for connected inhalers. Connected inhalers that only capture medication usage information have fit perfectly within the asthma digital ecosystem by providing key usage information and have been proven to increase days on treatment and reduce asthma attacks for patients.
The commonality between inhalers and pen injectors is that they are both multidose devices. An inhaler can be used for up to a month. Pen injectors utilize cartridges with formulations designed for repeat use. Each time the pen injector user uses the device, they attach a new insulin needle. The pen injector and its drug typically are designed for up to a month of repeat use and are replaced afterward, similar to an inhaler. In such a scenario, having Bluetooth connectivity for both the connected inhaler and connected insulin pen injector makes complete sense. The user will pair the inhaler with the add-on or pen injector with connectivity to their smartphone whenever they replace their medication. The smartphone, with its processing capacity and user interface, is the focal point for patient end-data gathering and the central patient interface. For connected inhalers and pen injectors, connecting the user’s smartphone with the connected device does not take place when they need to take their medication but instead occurs when the user replaces their medication, typically at a scheduled time of the month. As such, performing the pairing operation and checking for proper connectivity between the inhaler/pen injector and the smartphone does not take place during the stressful act of taking the medication. On the other hand, prefilled syringes, prefilled syringes with needle safety devices, and autoinjectors are single-use ready-to-use devices designed to be discarded after use. Yet what we have seen is that many connectivity solutions put forth for these single-use devices follow the Bluetooth connectivity model of multi-use devices like inhalers, without taking into consideration the patient’s use situation or the disposable/single-use nature of the device. As a result, those solutions, if assessed at all, are guaranteed to be rejected by pharmaceutical manufacturers.
Above, we discussed the connectivity technology used. The choice of wireless communication technology is one of the essential early considerations for a connected device. It needs to be selected based on the device’s intended use situation, as well as with the end user in mind. We’ll continue with the device’s use situation just alluded to and will utilize our SureMed Tech’s product lines to demonstrate why, when selecting the wireless communication technology, we must also take a holistic view of the end user for the developed connected device to be successful.
Use NFC, Not Bluetooth, For Single-Use Disposable Injectors
We at SureMed Tech believe disposable connected injectors such as digital syringes and autoinjectors need to utilize NFC as opposed to Bluetooth, based on the product’s use situation. Only fixed-dose drug products are placed in disposable injectors such as prefilled syringes or autoinjectors as they are injected in full and disposed of afterward. As such, the patient will only take those products out of refrigeration at the time of use. If the digital solution for these disposable injectors is built around Bluetooth, as many failed injector digital connectivity products have been in the past, following the connected inhaler model, what we are essentially doing is complicating an already complicated process for the user by asking them to perform Bluetooth pairing and check for successful Bluetooth pairing while they are trying to keep straight all the medication steps while mentally preparing for the injection. This adds to the user’s stress and anxiety. With NFC, there is no need to perform Bluetooth pairing; it is ready to go for the patient. Their preparation steps stay the same as before, and only after completing the injection do they just tap their used injector device to their smart phone to record that they have taken their medication, similar to tapping their phone to pay at the grocery store or tapping their phone to open a hotel door.
While there may be other wireless communication technologies that allow for quick pairing, such as QR codes and smartphone camera mode, those still introduce additional steps and adds complexity to an already complex process. Over time, the user will find it cumbersome to use the smartphone camera. We found in our interaction with patients that over time, patients feel it is more like proving that you have done your homework as opposed to being proactive with their medication tracking. Second, except for online banking, we are not in a habit of using the camera feature to do anything other than taking pictures or videos.
Taking into account our everyday experience, NFC will prove easy to adopt and most beneficial to the patient. The reasoning is that NFC is widely used, from credit cards to hotel room access, company IDs and access badges, metro and train station passes, and smartphone tap and pay functionality. As such, NFC and its tap-to-use is ingrained in our everyday behavior. This is important when building digital products as we need to utilize behaviors and technologies that are entrenched in our daily lives to minimize adoption challenges and ingrained behavior incompatibility. Imagine if a digital product is built on ZigBee wireless communication; it may be the best product and ZigBee may provide it with the best wireless communication for its need. Nevertheless, since no product on the market uses ZigBee, it would be a hard ask for the end user to learn how to use ZigBee just to use your product.
Consider The Needs Of Your Users
While matching the right wireless technology to the end use situation and the end user’s preconditioned experience and behavior is an essential step, in addition we must also consider the various stakeholders needs within a digital ecosystem. In the case of healthcare providers and doctors, as mentioned before with the watch and car analogies, for a connected drug delivery device, the balance of information captured and the connectivity purpose should always be considered. Over the years, we have seen many products try to do more than needed and, in doing so, rendered the connectivity solution flawed and unusable as those additional features often result in products that provide distracting and sometimes counterproductive information. From all of our stakeholder interactions, we found that the purpose of the connected drug delivery device, be it accessory or integrated add-on, must only do one job and that is to provide a way for the patient to confirm to the digital ecosystem that they have taken their medication and nothing more. The digital ecosystem has other integrated solutions/products to provide the reminders, prompt for diary entry in the case of clinical trials, etc. Too often, we seen connected injector solutions try to perform more than is necessary, such as having a pre-injection login step that then walks the patient step by step through how to use the device or that provides a digital set of the product name, batch record, and/or expiration data information for the app to asks the patient to confirm against the syringe label. What’s worse is that we have seen some products that force the patient to go through a step-by-step walk-through of the device preparation and injection steps with each step requiring the user to tab a “next” icon on the smartphone that appears only at the end of each step tutorial.
This is all happening during or concurrently with the patient’s preparation and injection process, adding additional unnecessary anxiety and stress on the patient. In addition, walking the user through the injection steps should not be a forced activity every time the user gives themselves an injection. This is an onboarding feature that helps familiarize and/or remind the user of the administration steps. After the user has had experience, this should not be repeated every time. It would be like forcing the user to watch the same TV ad over and over and over. Instead, as we have advised countless pharmaceutical clients, this should be an optional feature, such as a help icon that the user can go to if they want a refresher on the steps association with taking their medication.
This gets into the other problem we have seen with failed connected injector solutions. The selection of connectivity technology is critical. To distinguish between an un-injected and an injected syringe or autoinjector, there are a number of designs that utilize a switch-based NFC design that, prior to injection has one signal and after injection has another signal. The problem here is that this design requires a manual switch interface (custom-designed atypically shaped finger flange that must interface with custom-designed atypically shaped plunge rod, for example), resulting in a complete change in product look and feel. We will discuss this shortly in this article. In addition, this also creates the unintentional problem that it requires the user to interface the device with the smartphone app once beforehand when the patient is preparing and getting ready to administer the medication and again after medication have been administered. This adds additional user steps and complexity.
Those designs are little better than Bluetooth as they still require the user to preregister the device – basically, pairing of the device to the app – followed by a second register or tap of the device to the smartphone after use. First, this creates a lot of room for user error and confusion, and this again asks the user to remember and perform additional tasks and procedures while they are preparing and getting ready for an injection. In addition, this is counterintuitive to our ingrained behavior. We do not tap our phone twice to pay at the cash register. We do not tap our ID badges twice, once to initiate and another tap to unlock the door. We are preconditioned through our everyday NFC-based products that one tap is enough. So, oftentimes we see patients with the aforementioned failed NFC devices at first tap the device to register/pair it with the smartphone, but then they do not tap the device again after injection. When asked why, the most common response is they forgot or that they thought one time is enough just like their badge at work. Similarly, when asked about the pre-use tap/pairing and post-use tap, participants typically find it cumbersome. Many participants, when asked what they think of using the two-tap NFC design in their daily lives, indicated they would most likely forget to do the first tap. When asked further, the reason given by a large number of participants is that with all the preparation, they would most likely forget to do the first tap and remember to tap the device to the smartphone when the injection is done.
Therefore, for adherence and compliance, it is essential to use a technology that only provides a signal after the device is used/after the medication is injected to confirm that the device has been used. And this is what we have done at SureMed Tech. We have developed our proprietary NFC technology whereby the device does not transmit any signal until it has been used, as shown in Figure 1 below. This way, it provides a much more reliable medication use confirmation. In patient surveys, we found that users prefer this type of connected device, as it is in line with their daily experience with NFC technology (badge access, tap to pay, etc.). In addition, we found that patients prefer a device that transmits signal only after use because it keeps things simple and straightforward for them…”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

18 Mar
FDA Releases Guidance On Digital Health Data Acquisition In Clinical Investigations
John Giantsidis, president of CyberActa, Inc, wrote in the current issue of meddeviceonline: “Increasingly, digital health technologies (DHT) are becoming part of the conduct of clinical trials for pharmaceuticals and medical devices. They cover an extremely broad range of applications, including ingestible and implantable sensors, wearable devices carried by patients to measure certain health related parameters, digital/remote monitoring of drug intake, electronic signatures on consent forms, health data analytics through processing data that support bioinformatics modelling, and electronic patient diaries. The options are increasing daily.
Consequently, the FDA is taking steps to deliver on its promise to make the U.S. the center for digital health innovation by issuing a draft guidance1 providing its recommendations to clinical trial sponsors, investigators, IRBs, and others on the use of DHTs for remote data acquisition from participants in clinical trials evaluating drugs and biological products, medical devices, and combination products. A word of caution: It is crucial for clinical trial sponsors to understand and evaluate whether their DHT meets the definition of a significant risk device under 21 CFR 812.3(m) and as such would require submission of an IDE application to FDA under Part 812 for the same clinical investigation. Equally important, sponsors would have to determine whether the DHT is a medical device (under EU MDR 2017/745) or in vitro diagnostic (under EU IVDR 2017/746).
Goals Of The Draft Guidance
FDA recognizes the large spectrum of DHTs available for potential use in a clinical trial. As such, the draft guidance outlines recommendations intended to facilitate the use of DHTs in a clinical trial and addresses some of the information that should be included in an IND or an IDE (investigational device exemption) application for a clinical investigation in which the sponsor plans to use one or more DHTs or in a marketing application that includes such a clinical investigation. The principal areas of the FDA’s draft guidance are:
- Description, selection, and rationale for DHT use in clinical trials
- Verification, validation, and usability of DHTs
- Evaluation of clinical endpoints and statistical analysis
- Risk and other considerations when using DHTs
- Record protection and retention
Description, Selection, And Rationale For DHT Use In Clinical Trials
FDA outlines the expectation that sponsors must include in their applications an explanation as to why the DHT is fit-for-purpose, and this fit-for-purpose justification is to be based on the clinical event or characteristic of the disease or condition of interest that is to be measured, the proposed clinical trial population, the design of the clinical investigation, and the characteristics of the DHT that may influence trial participant use. Sponsors would also have to evaluate whether the participant’s own DHT and/or general-purpose computing platform is appropriate to reliably collect or facilitate the collection of data during the clinical trial. It is important to note that while these points seem straightforward and clear, the draft guidance does not provide what level of evidence is required to be fit-for-purpose. As it relates to the selection criteria, FDA sets three overarching topics to be addressed in the fit-for-purpose rationale:
- Clinical Trial Population – Education, language, age, and technical aptitude to ensure that trial participants will be able to use the DHT as intended for the purposes of the clinical trial.
- Design and Operation of DHTs – Fit-for-purpose rationale must cover the following:
- Design (e.g., material, size, weight, appearance, portability) and ease of use
- Power needs (e.g., battery life, charging recommendations)
- Operational specifications (e.g., data storage capacity, frequency of data transmission)
- Alerts (e.g., low battery, poor signal, data not being recorded or transmitted to the server)
- Environmental factors (e.g., temperature) that may affect the performance of DHTs
- Availability and capacity of both clinical trial participant and sponsor network systems to handle the volume of data obtained from frequent or continuous recordings
- DHT privacy and security to prevent unauthorized access to the DHT and the data it collects.
- Use of a Participant’s Own DHT or General-Purpose Computing Platform and Telecommunications – The fit-for-purpose rationale must consider whether allowing trial participants to use their own DHTs reduces the burden on the clinical trial participant. FDA did not outline or define “highly specialized or customized measurements,” but those would require sponsors’ DHTs. In the submission, the sponsor would have to enumerate:
- minimum technical specifications (e.g., operating system, storage capacity, sensors);
- performance specifications (e.g., precision and accuracy); and
- brand, model, and/or version that meet the minimum technical and performance specifications.
Verification, Validation, And Usability Of DHTs
FDA outlines that sponsors must confirm, through verification and validation activities, that their proposed DHTs are fit-for purpose. However, the terms “verification” and “validation” as used in this guidance are not the same as in 21 CFR 820.3(aa) and 820.3(z) under the QSR for devices (21 CFR part 820) or the terms “device software function verification” and “validation” as described in FDA’s own guidance General Principles of Software Validation. According to the draft guidance, FDA uses the term verification in this guidance analogously to analytical validation2 and the term validation analogously to analytical validation and clinical validation.3 The draft guidance spells out considerations that would be expected in the submission regarding the validation and verification of DHT hardware, DHT software, general-purpose computing platforms, as well as interoperability of connected systems with the DHT and usability studies on the DHT. Be advised that the FDA’s expectations are that sponsors will include usability studies to confirm the suitability of the DHT and/or general-purpose computing platform for the proposed clinical investigation. The guidance establishes several criteria to be met:
- Enroll a cohort that is similar to intended trial participants;
- Test the ability of future participants to use the DHT as directed in the trial protocol; and
- Assess whether users are able to enter all data before being logged out of a DHT.
The direct reference to the 2016 guidance on Applying Human Factors and Usability Engineering to Medical Devices indicates that the FDA would expect to receive from the sponsors human factors validation testing to demonstrate that the DHT can be used by the intended users without serious use errors or problems, for the intended uses and under the expected use conditions.
Evaluation Of Clinical Endpoints And Statistical Analysis
Of particular note is the proposed requirement to establish clinical endpoint or endpoints measured from data collected through a DHT, especially when justifying a novel endpoint using data captured by the DHT, where sponsors are to justify:
- Whether the endpoint is a clinically meaningful reflection of how a participant feels, functions, or survives;
- How the endpoint relates to other endpoints of effectiveness that have been used to support a marketing authorization for a similar indication; and
- Whether the novel endpoint is a sufficiently reliable measure of disease severity or health status.
Similar expectations are extended to the statistical analysis plan, where the definition of the endpoints and the source data from which the endpoints are derived for each trial participant and intercurrent events that may be related to the DHT should be prespecified in the statistical analysis plan. The listed intercurrent events are:
- Software updates that change how the data are collected or that change the algorithms used to process data
- Software incompatibility caused by operating system upgrades
- Trial participant error or noncompliance with study procedures using the DHT
- DHT failure
- Data transmission failure
Risk And Other Considerations When Using DHTs
FDA outlines three overarching risks to trial participants associated with use of the DHTs for data collection: clinical, privacy, and informed consent. It is not evident from the draft guidance whether such information is to be part of a submission or simply a documentation of FDA’s current thinking on the matter. Irrespective of the communication status, regarding clinical risks, sponsors are expected to:
- Analyze DHTs for physical features that could cause injury to participants;
- Evaluate likelihood of erroneous measurement, especially in situations where measurements provide feedback to modifications or changes in the administration of investigated products; and
- Assess any cybersecurity threats that may impact the functionality and safe data storage and transmission.
In furtherance of 21 CFR 50, the clinical trial risks and the requisite information on the informed consent process, the FDA emphasizes that the informed consent process must:
- Describe any reasonably foreseeable risks or discomforts to the subject, including reasonably foreseeable risks or discomforts related to the use of the DHT in the clinical trial.
- How the risks most likely to occur could be mitigated should also be considered for inclusion.
- Include a statement indicating that use of the DHT during the clinical investigation may involve risks to the subject that are currently unforeseeable.
- Explain the type of information that will be collected by the DHT, how that information will be used and monitored and the measures to protect a subject’s privacy and data, and the limitations of those measures.
- Specify who may have access to data collected through the DHT during or after the clinical investigation and during what time frame.
- Understand and map the impact of each end-user license agreements or terms of service as a condition of use because they may allow DHT manufacturers and other parties to gain access to personal information and data collected by the DHT. As such, sponsors and investigators should ensure that the informed consent process explains to subjects that their data may be shared by the DHT outside of the clinical trial, according to the end-user license agreement or terms of service.
Record Protection And Retention
FDA outlines that sponsors and investigators are responsible for recording and retaining data captured by the DHT in accordance with FDA’s record retention regulations for sponsors and investigators. It also makes reference to another draft guidance, “Use of Electronic Records and Electronic Signatures in Clinical Investigations Under 21 CFR Part 11—Questions and Answers.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

11 Feb
Nine Pitfalls To Avoid In Data Integrity
Peter H. Calcott, D.Phil., president and CEO of Calcott Consulting LLC wrote in the current issue of meddeviceonline:
1. Part 11 Compliance
Part 11 compliance has been around for about 20 years,1 but still today I see confusion in the industry. While most elements have been implemented successfully, in part attributable to the array of great software available in the marketplace, there are still areas where people stumble. I will illustrate two areas.
First, while almost all my clients purchase software in the marketplace, the systems fall into two types for the purpose of this point. The systems that reside in the cloud, where you access the application via the internet, usually are very robust. However, I have seen clients disable certain features that can have impact (e.g., audit trails). On e-systems that reside on a PC at the site of use, I have witnessed several problematic incidents. These all focus on the integrity of the clock used to date-stamp data. In these examples, the software has used the Microsoft (computer) clock to date-stamp the data rather than the software clock. In both cases, the Microsoft clock was not protected, and I could change the date and time with a click of the mouse. This rendered the date stamp worthless. The simple fix was for the administrator to lock the computer setting so that analysts could not change it. Of course, the system administrator can still do that.
Second, assignment of appropriate access based on job function is critical for meeting Part 11 compliance. At larger companies, there are at least three levels of access, easily recognizable. I will use setting up access levels for a High Pressure Liquid Chromatography (HPLC) as an example. The most restrictive is for staff who simply review data, either rejecting or approving the results. This is common for supervisors or manager-level staff. The next level is assigned to analysts, where they need to be able to set up runs, review data, and make adjustments to, for instance, integration parameters. And finally, there is the system administrator, who has complete access to the inner working of the system. In some smaller companies, these assignments are often blurred, with “super users” having administrator rights although they actually run samples and process data. This means that these analysts can actually access the file systems and make major adjustments. This leads to situations where DI can be questioned. It is particularly important to assign the access based on job function and separate the administrative functions from analyst roles. This tends to be a problem particularly in smaller companies.
2. Integration Of HPLC Chromatograms
Ideally, software should be set up to run automatically and integrate correctly every time. However, in many analyses, integration is not perfect, requiring post-analysis adjustments. If this is the case, it is paramount to incorporate into the method SOP a procedure to follow, with appropriate documentation, so the reintegration is performed in a reproducible compliant and documented manner. Without these controls and checks, it leaves the company open to DI questions.
3. Out Of Specification (OOS) Investigations
Even after the “Barr Case” of 1993,2 companies run into problems with how they conduct OOS investigations. There needs to be a robust SOP detailing how you proceed when a suspected OOS is encountered. Initially, before any investigation into product quality, there needs to be an assessment as to whether the method was run correctly in the laboratory. If an error can be demonstrated, the whole result or even the whole run can be nullified and the run or sample repeated. Once the run is shown valid, or at least cannot be nullified, then a product investigation can be considered. A detailed investigation plan, including repeat testing or retesting, must be drawn up and executed. Failure to follow a structured plan can create doubt about your DI status and conclusions.
4. Environmental Monitoring (EM) Data
Particularly in sterile or aseptic processing operations, many EM data are generated and analyzed. Obviously, the correlation between numbers of colonies on plates and the results on test forms must be perfect. So, when I routinely audit operations, I often review these forms. Even well-run operations will pick up counts on plates quite normally. If I see page upon page of zero colony forming units, my suspicions are triggered. If it looks too good to be true, it usually is.
5. Reports That De-emphasize Data That Compromises The Study – Validation And Investigations
As I indicated in part 1 of this article series, any discrepancy or deviation or anomalous data that is generated in an investigation or validation must be considered in the context of the end result of the report. Too many times, I have found results that might cast doubt on a conclusion are ignored and not discussed. Not all “failing” results or deviations will necessarily nullify the conclusion. In many cases, other tests can be performed to address the anomaly. At the end of the day, you want a report that can be read by others (including an inspector) that is correct and convincing.
6. Cherry-Picking Data
In many MHRA and FDA presentations and guidances,3-7 they have described cherry-picking of data. That is the tendency to keep testing until you get the result you “want” – usually a passing result. It often manifests in using unofficial databases to house data, running trial samples, using test samples to “calibrate” systems, and the list goes on. In the GMP world of validated methods, you get a chance to run a sample once according to the SOP. Only if you can prove there was a lab error can you justify nullifying the test and repeating it. While FDA warning letters are rife with incidents, I have found in auditing it often happens in smaller companies that are transitioning from being solely a research company to a development company moving into clinical trial manufacturing. Often, the senior staff is research trained and not familiar with the GMP requirements. It is a hard transition to make in a career. I know because I made that transition a long time ago.
7. Making Data And Sampling Ports Accessible
Although I have never seen an example of this in my years of auditing, I am including it because the MHRA used this example in its 2018 guidance 6. By this, they mean that a sampler might be tempted to sample not from the correct port in, for instance, a WFI loop, but rather another from a more accessible one, if the former is difficult to get to. So, we must be vigilant to assure we do not install obstacles in the way of our staff getting their jobs done correctly.
8. QA-Issued Forms
Any blank form used in your operations (on the shop floor or QC labs) must be a controlled form. That is, it must be QA issued (appropriately reviewed and approved) and be issued with a unique identifier. QA needs to keep an inventory of those issued, when, and to whom. If the forms are available online and can be printed off by an operator, then the control is lost. A form can be filled in or destroyed with no record. I have found this is a difficult principle for some smaller companies to grasp. If you do not track issuance of your forms, you are open to questions about the integrity of your documentation.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

15 Jan
Advancing Patient Care With Wearable Medical Devices
Sara Cinnamon, Amy Hung, Tobias Silberzahn, and Eli Weinberg (McKinsey) wrote in the current issue of meddeviceonline: “While wearable medical devices offer the opportunity to monitor, diagnose, and deliver individualized care to patients, adoption by clinicians has yet to gain significant traction. However, the COVID-19 pandemic has raised awareness of wearables and how they can provide clinical benefit in a remote setting. Many medical practitioners and patients were introduced to new ways of delivering care, including expansion of existing telemedicine and remote patient monitoring.
Traditionally, mainstream wearables familiar to consumers were not developed with clinical rigor from the outset. Medical devices have a high bar for accuracy and validation to integrate and deliver the type of data required for clinical use within the healthcare delivery ecosystem. Consumer devices often leverage less-accurate sensors or inferred measurements to support informational and non-medical feedback to the user. Seeing the evolving remote care trends and revenue potential of clinical use, consumer device makers have begun closing that gap. For example, Apple has received FDA clearance for the ECG function on the Apple Watch, opening the door for its use in clinical settings. Oura, maker of a smart ring that senses activity, sleep, body temperature, and heart rate variability, has partnered with UCSF to create an in vivo trial to rapidly collect correlative data between biomarker changes and COVID-19 diagnoses to inform a diagnostic algorithm.
In fact, research shows the wearables market is estimated to grow considerably, with 19 percent CAGR from 2020 to 2030, representing potential economic value of $0.5-$1.8 trillion by 2030. This growth includes consumer, industrial, and medical applications, but market activity indicates significant promise (and movement) in the healthcare domain:
- In 2020, global shipments of hearables, watches, wristbands, and other wearables stands at 444.7 million units
- Investors have committed more than $400 million to wearables-focused startups since 2015
- Large medical device companies are acquiring smart devices, such as Stryker’s acquisition of OrthoSensor and Boston Scientific’s acquisition of Preventice Solutions
- Academia continues to drive innovation in ever smaller technology, with advances in implantable chips from UC Berkeley, flexible batteries from Stanford University, and flexible sensors from the University of Illinois at Urbana-Champaign
Device manufacturers are well positioned to capitalize on this unique opportunity. However, to deliver devices useful for clinicians and patients, wearable technologies must advance further. Breakthroughs in healthcare will likely come from devices that not only accurately measure disease-specific biomarkers at home but also provide real-time contextual data on other features that affect disease management. These insights must be shared via a simple user experience to encourage patient adoption and must demonstrate improved medical care and reduced costs to providers and payers.
Based on our experience, wearable device manufacturers and service providers should focus on three areas to ensure success in the market:
- Meeting and exceeding user needs
- Delivering rigorous, compliant data and insights for the entire patient care ecosystem
- Crafting a viable business model
Here, we provide an overview of each focus area. In future articles, we’ll dive more deeply into each.
1. Meeting And Exceeding User Needs
Patients and clinicians expect sufficient long-term benefits in exchange for using wearable devices, what can be called “return on engagement.”
For patients, return on engagement translates into benefits such as reassurance that a professional is monitoring them and able to deliver care above and beyond what point-in-time visits can offer. They expect this to come with minimal discomfort from the ongoing use of these devices. If the device is uncomfortable or creates substantial burden on patients’ daily routines, they won’t use it. As a result, strive to develop smaller, less obtrusive devices with longer battery life. Device designers must also understand how physical and cognitive ergonomics affect device operation and data comprehension. Patient interactions with the device should be simple, seamless, engaging, and personal.
Clinicians’ return on engagement can be realized by integrating device read-outs into their current systems and workflows seamlessly, as well as by generating insights that they wouldn’t be able to infer otherwise. Devices and associated services that combine wearable data with real-world data sources to reveal patterns and trends will give clinicians a more comprehensive picture of patient health compared with point measurements in the doctor’s office, enabling more efficient disease management.
For both clinician and patient, safety, data security, and efficacy are table stakes, with correlation to better patient outcomes the ultimate sign of success.
2. Delivering Rigorous, Compliant Data And Insights For The Entire Patient Care Ecosystem
The data collected by wearables must be sensitive, specific, clinically validated, and offer more than that available through current standards of care. Such rich data will enable a better understanding of disease progression, more precise patient segmentation, and, ideally, better clinical outcomes.
However, the data alone won’t provide this level of value. Wearables must also deliver a level of contextualization and analysis that requires robust algorithm development hand-in-hand with device development (e.g., to accommodate individual variations and provide clear clinical insights). These insights must then be visualized and displayed in a way that is easy for users to comprehend and act upon.
Device makers must also consider the needs and requirements of other stakeholders in the patient care ecosystem, including payers, hospitals, and regulators. Data serves as the connective tissue that enables this ecosystem to better work together for the good of the patient, making it critical that these stakeholders can share and communicate the data and insights seamlessly. Industry standard protocols such as FHIR or HL7 enable interoperability to ensure all stakeholders have access to the right information at the right time. As a result, consider how your device’s data will be transferred to the electronic health record for clinician review or how it will interact with third-party APIs.
Finally, data storage and transfer must comply with industry regulations — such as HIPAA, GDPR, PIPEDA, and CCPA — and best practices for privacy and security. For instance, use industry-standard encryption for data on devices and during transmission, and prevent or mitigate unauthorized access to devices, smartphones, and servers. As regulations evolve, such as with the European Commission’s recently announced legal framework for trustworthy AI, so must manufacturer approaches.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801
MedSysCon – The faster way to medical device compliance
The regulatory and quality landscape for medical devices is constantly evolving. The mission of MedSysCon is to deliver specialized consultancy services for regulatory affairs and quality assurance, focused on the digital health industry.

For further information please get in touch with us:
+49-176-57694801
Make Your Medical Device Start-Up More Investable
Jim Kasic, founder and chairman of Boulder iQ, wrote in the current issue of meddeviceonline: “Say “start-up” in the medical device community and the conversation can move quickly to funding. Who will invest? How much? How long will it take?
A particularly tough challenge for medical device start-ups is that the executives tend to be the scientists and inventors of the device. When it comes to funding, you need to trade your inventor hats for business ones and learn to think like an investor. It’s not an easy task, and with most medical device start-up companies operating in a very competitive landscape, the need for speed in attaining this business acumen is critical.
How to do it? Here are five tips to help a medical device start-up be more interesting, more attractive, more investable – and less risky – to potential funders.
1. Define your market.
In other words, do your competitive homework. As wonderful as you believe your device is, make sure you have something new, something that performs a function at a lower cost than existing devices, or offers more for the same cost. If you can prove this, and communicate it clearly, you may have an attractive investment.
A market that is eager for a device to answer a clear unmet need is the optimal “defined market.” Investors want to see that your device fits clearly into a specific medical treatment area or diagnostic process and that it enhances or replaces current procedures. In the medical device start-up world, there is no place for “missionary” sales work, where you must convince a customer to believe in a new product concept – especially one that requires a change in current procedures. This process, which is incredibly difficult to carry out, also is long and involved – none of which works for an investor looking for timely returns.
- Create a device that will be used in procedures that are already being performed.
- Make sure the device will be used in a specific application.
- Define the physicians or other clinicians who know they need a new or improved way to perform the procedure.
- Calculate the market size. This could be based, for example, on the number of specific surgeries conducted each year that will use the device, or other relevant use factor.
- Know how to identify where the users are and how to reach them.
2. Make sure that your market will be willing to accept and pay for your device.
Investors want to make sure that buyers will be willing to accept and pay for your device. So, while medical device developers all want to create devices with clinical benefits, they must also have economic benefits.
Once ready for market, the reality is that a purchasing agent must deem your device necessary and affordable. Even if a physician requests it, the purchasing department has to make sure that the device satisfies the physician’s need at the lowest cost. One way to help assure this acceptability is to demonstrate that your device can be paid for within current reimbursement structures, specifically through Diagnosis-Related Group (DRG) reimbursement and Current Procedural Terminology (CPT) codes.
A DRG reimbursement is associated with a patient’s diagnosis, which typically is associated with a fixed dollar amount of reimbursement – i.e., a DRG reimbursement. The healthcare provider is expected to treat the diagnosis for this fixed amount. If your device can help them do that for less money, you may have an advantage in that provider’s system.
CPT code reimbursements are associated with a physician’s specific skills and their use of equipment and tools to perform a particular procedure. For example, physicians performing cardiac catheterization procedures require special training and qualification. The CPT code provides additional reimbursement for the special skills and equipment necessary to perform this treatment. Medical device entrepreneurs should understand that obtaining new CPT codes is a lengthy process typically requiring two or more years, along with several peer-reviewed publications attesting to a device’s advantages. However, if your device fits within existing CPT codes, you will have a clear economic advantage in investors’ eyes.
3. Set a price that supports high margins.
Investors are looking for a solid story of why you can expect high margins and how you will maintain those over a reasonable period of time.
Medical device values are based more on the importance they bring to a particular procedure than on manufacturing costs. Devices that offer a truly unique market advantage and provide excellent value to each of the “3 Ps” (the patient, the physician, and the payer) are in the best position to support high margins.
Gross margin is calculated as:
(Average sales price – Cost of goods sold) x 100
Average Sales Price
Gross margins above 70% are typically attractive to investors. A gross margin in the mid-80% range offers excellent returns. Investors also understand that these high gross margins are needed to support the overhead costs of working and innovating in a regulated industry.
Investors will want to see pricing that is commensurate with those margins. Think of it this way. In the early and mid-1900s, there was such a thing as “penny candy.” It was fun for kids to buy, but no matter how high the gross margin was on a piece of candy, the manufacturer was never able to make a significant amount of money on that product alone. The selling price was just too low.
In the medical device community, developers can look at the number of procedures performed each year that would employ their devices. Very few medical procedures are performed to the tune of millions per year, but it’s very possible that a device would be needed for hundreds or tens of thousands of cases per year. If so, there’s likely an opportunity to make a device that can be produced in a large enough volume to support high margins and command a price that will provide sufficient returns to investors (and the developer).
Setting sales prices encompasses far more than the cost of making them and obtaining required regulatory and quality certifications. Medical device developers may overlook the real costs of marketing and selling as they analyze margins and pricing. Consider that there is substantial cost in communicating to potential buyers and convincing those buyers – usually physicians and payors – to purchase the device. You must balance the cost of obtaining a customer with your gross margin and number of devices a customer is going to buy.
For example, you want to make sure that the margins on the device can cover the sales and marketing costs. Imagine a device is sold for $2,000 with an 80% margin – meaning the company would make $1,600 on the sale. But that’s before overhead costs that include sales and marketing. If those costs are $5,000 – not unrealistic – then the company will not make any profit, and in fact, will lose money on the deal.
However, if a customer buys four devices, the company would be making a total gross margin of $6,400. Subtract that $5,000 cost for sales and marketing, and now you are covering the cost of converting the prospect into a customer as well as starting to create a positive cash flow.
Conversely, let’s say you have a less expensive device that sells for $1,000. At that price, you’ll need to sell twice as many to cover the cost of converting a prospect into a customer. The bottom line is that start-up device developers must do the math, be realistic, and understand the importance of pricing to investors.
4. Confirm your FDA pathway.
Regulatory hurdles can present some of the greatest risks for investors interested in start-up medical device businesses. Sadly, horror stories abound about companies that were convinced they had a clear FDA pathway and later found otherwise. Hopeful investors and investment dollars went by the wayside.
For example, a client of ours was developing a drug delivery device, which it considered a simple pumping or transfer mechanism. Far along in the product development cycle, they learned that the FDA classified the device as a “combination product” and required proof that the drug container provided long-term stability for its contents. As a result, instead of looking to obtain a 510(k), the company faced lengthy, expensive drug storage testing to confirm that the device had no adverse effect on the drugs it would deliver.
Other examples include devices that try to combine predicates in a 510(k) application, when the devices actually have separate indications of use. Instead of a 510(k), a lengthier and more involved de novo application might be required.
Medical device start-ups should also be aware they may be required to conduct clinical trials to help establish a device’s safety and efficacy. They would then need to submit a Clinical Evaluation Report (CER) with appropriate statistical evaluation of the results with their 510(k) application. Some 510(k) devices are cleared without clinical data requirements, but not all. To assume that no clinical data will be required can lead to a grossly underestimated timeline and budget, which translates to poor investor returns. The best advice is to check it out in advance, consulting with industry experts or the FDA if needed.
The bottom line is to never assume a simple path to regulatory clearance. While some cases may be straightforward, it is almost always advisable to take the time to get an opinion from a regulatory affairs professional or ask the FDA to classify your device by submitting a 513(g) application.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

09 Nov
Meet Hearables – The Next Revolutionary Medical Devices
Hearables – The Next Revolutionary Medical Devices
A conversation with Jacob Skinner, Ph.D., CEO, Thrive Wearables presented in meddeviceonline.com gave insights into a new category of medical devices: “
Hearables, in-ear devices that use sensors to monitor health, are on the brink of becoming the next revolutionary technology. While in-ear devices have been around for decades in various iterations (used primarily for transmitting sound), the application of this technology in the medical field is a relatively new area to explore. Jacob Skinner, CEO of U.K.-based wearable technology design and development consultants Thrive Wearables, discusses its possibilities.
Why Are Hearables Becoming More Important As A Technology Now?
Skinner: In recent times, sensor technology has shrunk significantly so that all kinds of capabilities can be added to in-ear devices, creating a subcategory of wearable technology that we call hearables. Additionally, most of us carry a supercomputer around in our pockets, which can process data sent to, and received from, in-ear sensors.
JacobTechnology for transferring information is getting better, too. Early Bluetooth headphones traded audio quality for convenience, but today’s wireless earphones are good enough for all but the most dedicated audiophile. Last year, a new standard, Bluetooth LE Audio, was announced that can support hearing aids, streaming to multiple devices, and the Low Complexity Communications Codec (LC3, which transmits at lower bit rates, meaning that the device can use less power and can be less bulky).
In addition, almost all of us use earphones of some kind, whether they are wireless, Bluetooth-connected, in-ear buds, or wired headphones that go over the ear. We’re all pretty comfortable with the technology, which helps with patient adherence of this burgeoning field. So, the reason hearables are growing now is a combination of a form factor that we’re all used to and improvements in technology that enable us to make earphones do more than ever. It’s common for wireless earbuds to have a microphone, for example, and a sensor that stops playback when an earphone is removed from the ear. But we are now seeing them become health and fitness devices.
What Can Hearables Bring To The Health And Wellness Space?
Skinner: Some of this technology will simply offer an improved listening experience, based on individual circumstances, and some products are merging traditional ear device uses (listening to music or phone calls) with medical uses. EVEN’s headphones and Nuheara’s IQbuds, for example, both adapt their output to boost frequencies the listener doesn’t hear so well. Everyone’s hearing is slightly different, so compensating for personal limitations or age-related hearing loss can help.
Other technologies use earphones to monitor activity, stress, and even the brain. Sensors can be used for photoplethysmography (PPG), which is the use of light to detect changes in blood volume. That can tell you things like oxygen saturation (SpO2), blood pressure, and pulse rate. Blood pressure is a good measure of overall health, while SpO2 can monitor lung conditions, sleep apnea, and patients with cardiac conditions, among other things. Analyzing changes in this data could provide early indication of heart or blood pressure problems, as well as things like stress or drowsiness.
We can measure heart rate variability, too, which is the time difference between heart beats. A heartbeat of 60 beats per minute is not literally one beat every second. The gaps between beats will vary and the greater the variability, the better condition you are in. Using an in-ear sensor to measure this can help track overall fitness or recovery from a heart procedure.
Another way to monitor heart activity is with electrocardiography (ECG) sensors, which track electrical impulses, while electrodermal activity (EDA) sensors can be used to analyze breathing patterns. Electroencephalography (EEG) sensors, like those in the Kokoon sleep headphones, use electrical signals to monitor brain activity; Kokoon uses the information from its sensors to play white noise for those with trouble sleeping (Kokoon recently won a Red Dot Award in the Product Design category from Red Dot GmbH & Co. KG). Brain monitoring is developing rapidly and could be used to monitor stress, epilepsy, or even complex mental illnesses.
These are far from the only options. Temperature, motion, and other metrics can all be gathered from hearables. Electrical signals can track eye movement, which offers a way to monitor attention and alertness. Measuring changes in the shape of a user’s ear canal can identify facial expressions, which could become a means for controlling these devices or play a role in mood tracking.
What Can We Learn From That Data?
Skinner: Hearables can be useful for telling us what is happening in the body right now. They could tell you whether you are within the correct heart rate range for your exercise plan, for instance, and use an audible signal to tell you to speed up or slow down.
But trend data can be even more useful. Once a system begins to understand what is normal for your body, it can use anomalies to detect potential health problems. Combining several measures, along with things like voice recognition and head movement, could be a powerful indicator of overall health and well-being. Abnormal data could trigger anything from recommended deep breathing exercises for stress relief to scheduling an appointment with a clinician based on emerging cardiac symptoms.
When many people use these devices, their data can be analyzed in aggregate – with appropriate privacy controls. That would allow artificial intelligence and analytics to look for patterns and draw conclusions beyond those that can be derived from individual data. We might identify new warning signs of heart disease, for example, based on data collected from millions of people before they showed any symptoms.
Many of these applications are some time away from being reliable and widely used, but the pace of innovation is fast and increasing. We are just a few years away from widespread consumer use of hearables for health monitoring, but specific, niche medical uses are already happening.
Why Are Hearables Ideal For Data Collection Rather Than Wrist-Based Wearables, And What Are The Technological Challenges?
Skinner: Wearable tech for health and wellness has so far focused on wrist-based devices. However, signals from the ear are as much as 100-times clearer than those from the wrist. The inside of the ear is dark, closer to the body’s core, and the arteries are nearer the surface of the skin, which makes heart rate monitoring easier.
On the wrist, in contrast, sensors must work around muscles and tendons. During exercise, people tend to move their wrists a lot, while their heads stay relatively stable. Wrist-based monitors shine a light into the skin to detect blood flow, but if the device isn’t fitted snugly to the wrist, light can get in and reduce the effectiveness of the sensor. And unlike the inner ear, the wrist gets sweaty, which can make it hard for sensors to capture a good signal, resulting in variable quality insights.
The ear is also a good location for monitoring signals from the brain and eyes, and is less obtrusive than brain-computer interfaces that must be worn on the head. Speech recognition is easily handled by a microphone on an earpiece, and head movement is easily tracked, too.
The challenge with a device that you wear in your ear is that it has to be small and light, which obviously restricts how many sensors you can pack in. The sensors have to be small and there has to be room for a battery to power them. For a consumer device, you need to fit in speakers and a microphone, too. It’s a design challenge to make something that is comfortable to wear, with a practical battery life and the features that provide value. However, since the pioneering work of the German startup Bragi, a lot of investment has gone into creating new technologies that are specifically designed to work at ultra-low power and in extremely constrained form factors. There is also historical work in the hearing aid space, and this level of advancement has meant that hearable technology is able to move at significant pace. With this backdrop, and due to the consumer pull and market validation of AirPods, new sensors and advanced technologies are finding their way into our ear-worn wearables. These factors somewhat mitigate the challenges we face in product development.
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

31 Oct
Wearable Cardiac Monitoring
Tudor Besleaga, Ph.D., and Jacob Skinner, D.Phil., Thrive Wearables, wrote in the current issue of meddeviceonline: “Cardiac monitoring is almost a standard feature of modern wearables but there are still opportunities to get better information and do more with the data. Seizing those opportunities could transform everything from medical care to athletic training and general well-being.
Cardiac monitoring has been around for over 100 years, but improved accuracy and wearable technology mean we can now get a better view of cardio performance not only in the hospital but also in the home, at work, or even while the wearer runs a marathon.
Turning this monitoring into actionable insights promises many benefits. Can we determine how well someone is recovering from surgery? Can we measure stress? And can we do so with a system that is low on user friction and high on user value? At present, the most accurate measurements come from the most cumbersome devices, and devices that are comfortable to wear tend not to offer the richest data.
The opportunity for the next generation of products and services is to further blur the distinctions between devices designed for medical professionals, performance athletes, and consumers. We have some obstacles to overcome before we get there, though.
Why Monitor More Than Heart Rate?
It’s worth starting by asking why we want to monitor heart signals with wearables in the first place. It is routine to monitor the heart dynamics of all kinds of patients. The cardiovascular system reveals how a patient is recovering from surgery, the effect of drugs, and more. Sudden changes can indicate serious problems.
Innovation in high-quality cardiac monitoring has also come from devices for athletes. For example, heart rate variability (HRV) – the change in the time between heart beats – and resting heart rate are both indicators of overall fitness, so monitoring of these metrics first appeared in wearables for professional athletes and serious amateurs. These filtered down into more affordable consumer devices pitched as more general lifestyle and well-being assistants.
As well as cardiac monitoring for overall health, some devices can warn of sudden changes in heart rate intervals or test for atrial fibrillation (AFib), a common but potentially dangerous heart abnormality. In a sign of how serious diagnostic tools are being integrated into consumer products, the Apple Watch has been approved as a medical device by the FDA in the U.S. and by other regulators.
ECG Versus PPG
Different use cases will have different ideal technologies and form factors. Essentially, though, wearable cardiac monitors rely on two technologies with different approaches and distinct strengths and weaknesses.
The first is the electrocardiogram (ECG), first developed in the late 19th century. This measures the electrical signal produced by the heart as it beats and is a good guide to heart function. Measurement can be cumbersome, however. A hospital might use a 12-lead ECG, but this restricts movement and needs professional assistance. The consumerization of these devices sacrifices some data resolution but allows them to be worn unobtrusively and comfortably on the wrist for spot measurements instead of continuous ones. In recent years, some smartwatches have added ECG sensors, primarily for detecting things like AFib. These work by sending an electrical signal through the body so the user must wear the watch on one hand and touch the case with the opposite hand to complete the circuit.
Outside the hospital or a testing lab, a chest strap ECG is the best option. This isn’t as accurate as the multiple lead approach but almost everything in this space is a trade-off between user friction and perceived value. The value here is mobility, but the friction comes from reduced accuracy. Moreover, most people don’t want to wear a chest strap for long periods. There isn’t enough value. Cardiologists have tried to get patients to wear all kinds of devices and, other than implanted ones, devices on the wrist are about the only acceptable form factor for most patients.
That works well if the wearer can remember, or be reminded, to perform the ECG regularly but most people would prefer unobtrusive background monitoring. That can be done with a photoplethysmogram (PPG), which was invented in the 1930s and uses a light shone through the skin to track blood flow. This offers insights into cardiac hemodynamics, such as blood pressure trends and cardiac output but also provides localized measurements, such as tissue oxygenation. A downside of this type of sensing is its susceptibility to movement-induced signal loss, particularly at the wrist, where it’s most commonly worn. However, the wrist is a good location for user experience and PPG sensors are now standard in fitness bands and smartwatches, also making the sensor price point increasingly low.
PPG and ECG can complement each other in monitoring the cardiovascular system. The former can be adapted to various form factors and provides long-term continuous measurements, while the latter is limited to chest-based, albeit accurate, continuous measurements and wrist-based spot measurements. It is not uncommon to see PPG watches monitoring alongside chest straps during periods of intense exercise. Or PPG watches using ECG watch spot measurements to obtain quick but reliable and easily comparable readings.
Again, there is a trade-off. A PPG sensor might be most comfortable on the wrist, but the wrist isn’t the ideal place to monitor blood flow. We move our arms, especially when we exercise, which disrupts the signal and means lost data. The system must estimate, or do without, the missing information.
The Next Generation Of Cardio Sensors
Sensors, particularly PPG sensors, are improving all the time, so we can be optimistic about future devices. Most manufacturers have iteratively improved the designs of their PPG sensor modules regularly in recent years, indicating the likelihood of more application options as they improve further.
There are off-the-shelf integrated solutions for measuring PPG or ECG and exporting the data via a serial bus. However, these commonly fall short in signal quality compared to the best in class. Most developers design their own sensor systems, by combining a sensor-body interface, analog front end (AFE), and microcontroller unit (MCU).
The sensor-body interface, which is either an optical front end (OFE) for PPG or electrodes for ECG, is where the most value could be added. The OFE consists of one or more light emitters, commonly LEDs, one or more light sensors, commonly photodiodes (PDs), and a mask to control the light between them. The mask must ensure light passes through the tissue, rather than reflecting straight to the PD, that minimal ambient light enters the sensor, and that good pressure is maintained against the tissue.
The sensor-body interface must be optimized for where it will measure. Red or infrared light is fine for a fingertip PPG, for example, but green light makes data from the wrist less susceptible to motion noise. Some chest devices use both green and red to assess pulses at different depths; the higher the wavelength, the deeper it penetrates.
It is not uncommon for sensors to use discrete components assembled with the mask. However, integrated solutions can save space and cost. These provide PDs with peak sensitivity at the same wavelength as the LEDs and can output three channels (green, red, infrared). One downside is that the distance between the light emitter and the receiver is fixed and typically optimised for wrist cardiac measurement applications. That distance might need to be increased for measuring higher up the arm or at the chest in deeper tissue.
AFEs have improved a lot recently, particularly in signal preprocessing. They can measure ambient light and exclude the baseline, operate LEDs at very low duty cycles, increase energy efficiency, and filter specific frequencies. Some even calculate heart rate intervals for HRV. Many off-the-shelf integrated circuits (ICs) now provide collections of PPG, ECG, or both via multiple channels. When measuring ECG, it is important to select an IC that is optimized for dry electrodes, whether polymer conductive ones, as in chest straps, or stainless steel, as in many watches.
It is important to establish before development how much processing and analysis of the raw signal will be done on the device versus on servers. A lot of memory can be saved if only specific metrics are required, such as HRV, rather than complete raw data.
Since these signals are very susceptible to movement-induced noise, it is common to record accelerometer or inertial measurement unit (IMU) data along with the cardiac data. Motion data can help identify corrupted signal intervals and improve battery life by turning off recordings during high-intensity motion.
For ECG recordings, the MCU will be the main power consumer. Integrated solutions (OFE, AFE, and MCU) do exist. However, they are not quite plug and play, as different form factors require optimization of sensor position and body interface. Furthermore, the onboard MCU means higher energy consumption. They also share limited processed data, as raw sensor data exposes these companies to intellectual property (IP) leak risks.
Medical device manufacturers could benefit from pursuing a hybrid agile approach when developing devices with cardiac sensors, as iterative sensor implementations are to be expected.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

13 May
AI in MedTech – Risks and Opportunities
Risks and Opportunities of Innovative Technologies in Medical Applications
Dr. Abtin Rad, Global Director Functional Safety, Software and Digitization, TÜV SÜD Product Service GmbH wrote in the current issue of MedTechIntelligence: “An increasing number of medical devices incorporate artificial intelligence (AI) capabilities to support therapeutic and diagnostic applications. In spite of the risks connected with this innovative technology, the applicable regulatory framework does not specify any requirements for this class of medical devices. To make matters even more complicated for manufacturers, there are no standards, guidance documents or common specifications for medical devices on how to demonstrate conformity with the essential requirements.
Introduction
The term artificial intelligence (AI) describes the capability of algorithms to take over tasks and decisions by mimicking human intelligence.1 Many experts believe that machine learning, a subset of artificial intelligence, will play a significant role in the medtech sector.2,3 “Machine learning” is the term used to describe algorithms capable of learning directly from a large volume of “training data”. The algorithm builds a model based on training data and applies the experience, it has gained from the training to make predictions and decisions on new, unknown data. Artificial neural networks are a subset of machine learning methods, which have evolved from the idea of simulating the human brain.22 Neural networks are information-processing systems used for machine learning and comprise multiple layers of neurons. Between the input layer, which receives information, and the output layer, there are numerous hidden layers of neurons. In simple terms, neural networks comprise neurons – also known as nodes – which receive external information or information from other connected nodes, modify this information, and pass it on, either to the next neuron layer or to the output layer as the final result.5 Deep learning is a variation of artificial neural networks, which consist of multiple hidden neural network layers between the input and output layers. The inner layers are designed to extract higher-level features from the raw external data.
The role of artificial intelligence and machine learning in the health sector was already the topic of debate well before the coronavirus pandemic.6 As shown in an excerpt from PubMed several approaches for AI in medical devices have already been implemented in the past (see Figure 1). However, the number of publications on artificial intelligence and medical devices has grown exponentially since roughly 2005.
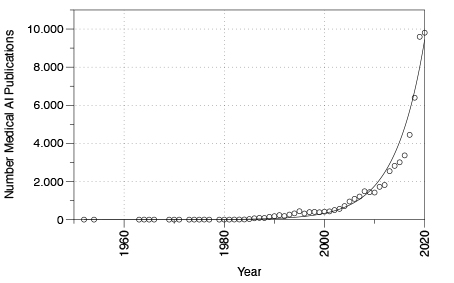
Figure 1. Selected PubMed publications for the search terms “artificial intelligence” and “medical technology” from 1950 to 2020.
Regulation of AI
Artificial intelligence in the medtech sector is at the beginning of a growth phase. However, expectations for this technology are already high, and consequently prospects for the digital future of the medical sector are auspicious. In the future, artificial intelligence may be able to support health professionals in critical tasks, controlling and automating complex processes. This will enable diagnosis, therapy and care to be optimally aligned to patients’ individual needs, thereby increasing treatment efficiency, which in turn will ensure an effective and affordable healthcare sector in the future.
However, some AI advocates tend to overlook some of the obstacles and risks encountered when artificial intelligence is implemented in clinical practice. This is particularly true for the upcoming regulation of this innovative technology. The risks of incorporating artificial intelligence in medical devices include faulty or manipulated training data, attacks on AI such as adversarial attacks, violation of privacy and lack of trust in technology. In spite of these technology-related risks, the applicable standards and regulatory frameworks do not include any specific requirements for the use of artificial intelligence in medical devices. After years of negotiations in the European Parliament, Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in-vitro diagnostic medical devices entered into force on May 25, 2017. In contrast to Directives, EU Regulations enter directly into force in the EU Member States and do not have to be transferred into national law. The new regulations impose strict demands on medical device manufacturers and the Notified Bodies, which manufacturers must involve in the certification process of medical devices and in-vitro diagnostic medical devices (excluding class I medical devices and nonsterile class A in-vitro diagnostic medical devices, for which the manufacturer’s self-declaration will be sufficient).
Annex I to both the EU Regulation on medical devices (MDR) and the EU Regulation on in vitro diagnostic medical devices (IVDR) define general safety and performance requirements for medical devices and in-vitro diagnostics. However, these general requirements do not address the specific requirements related to artificial intelligence. To make matters even more complicated for manufacturers, there are no standards, guidance documents or common specifications on how to demonstrate conformity with the general requirements. To place a medical device on the European market, manufacturers must meet various criteria, including compliance with the essential requirements and completion of the appropriate conformity assessment procedure. By complying with the requirements, manufacturers ensure that their medical devices fulfill the high levels of safety and health protection required by the respective regulations.
To ensure the safety and performance of artificial intelligence in medical devices and in-vitro diagnostics, certain minimum requirements must be fulfilled. However, the above regulations define only general requirements for software. According to the general safety and performance requirements, software must be developed and manufactured in keeping with the state of the art. Factors to be taken into account include the software lifecycle process and risk management. Beyond the above, repeatability, reliability and performance in line with the intended use of the medical device must be ensured. This implicitly requires artificial intelligence to be repeatable, performant, reliable and predictable. However, this is only possible with a verified and validated model. Due to the absence of relevant regulatory requirements and standards, manufacturers and Notified Bodies are determining the “state of the art” for developing and testing artificial intelligence in medical devices, respectively. During the development, assessment and testing of AI, fundamental differences between artificial intelligence (particularly machine learning) and conventional software algorithms become apparent.
Credibility and trust in AI
Towards the end of 2019, and thus just weeks before the World Health Organization’s (WHO) warning of an epidemic in China, a Canadian company (BlueDot) specializing in AI-based monitoring of the spread of infectious diseases alerted its customers to the same risk. To achieve this the company’s AI combed through news reports and databases of animal and plant diseases. By accessing global flight ticketing data, the AI system correctly forecast the spread of the virus in the days after it emerged. This example shows the high level of performance that can already be achieved with artificial intelligence today.7 However, it also reveals one of the fundamental problems encountered with artificial intelligence: Despite the distribution of information of the outbreak to various health organizations in different countries, international responses were few. One reason for this lack of response to the AI-based warning is the lack of trust in technology that we do not understand, which plays a particularly significant role in medical applications.
In clinical applications, artificial intelligence is predominantly used for diagnostic purposes. Analysis of medical images is the area where the development of AI models is most advanced. Artificial intelligence is successfully used in radiology, oncology, ophthalmology, dermatology and other medical disciplines.2 The advantages of using artificial intelligence in medical applications include the speed of data analysis and the capability of identifying patterns invisible to the human eye.
Take the diagnosis of osteoarthritis, for example. Although medical imaging enables healthcare professionals to identify osteoarthritis, this is generally at a late stage after the disease has already caused some cartilage breakdown. Using an artificial-intelligence system, a research team led by Dr. Shinjini Kundu analyzed magnetic resonance tomography (MRT) images. The team was able to predict osteoarthritis three years before the first symptoms manifested themselves.8 However, the team members were unable to explain how the AI system arrived at its diagnosis. In other words, the system was not explainable. The question now is whether patients will undergo treatment such as surgery, based on a diagnosis made by an AI system, which no doctor can either explain or confirm.
Further investigations revealed that the AI system identified diffusion of water into cartilage. It detected a symptom invisible to the human eye and, even more important, a pattern that had previously been unknown to science. This example again underlines the importance of trust in the decision of artificial intelligence, particularly in the medtech sector. Justification of decisions is one of the cornerstones of a doctor-patient (or AI-patient) relationship based on mutual trust. However, to do so the AI system must be explainable, understandable and transparent. Patients, doctors and other users will only trust in AI systems if their decisions can be explained and understood.
Differences Compared to the Regulation of Conventional Algorithms
Many medical device manufacturers wonder why assessment and development of artificial intelligence must follow a different approach to that of conventional software. The reason is based on the principles of how artificial intelligence is developed and how it performs. Conventional software algorithms take an input variable X, process it using a defined algorithm and supply the result Y as the output variable (if X, then Y). The algorithm is programmed, and its correct function can be verified and validated. The requirements for software development, validation and verification are described in the two standards IEC 62304 and IEC 82304-1. However, there are fundamental differences between conventional software and artificial intelligence implementing a machine learning algorithm. Machine learning is based on using data to train a model without explicitly programming the data flow line by line. As described above, machine learning is trained using an automated appraisal of existing information (training data). Given this, both the development and conformity assessment of artificial intelligence necessitate different standards. The following sections provide a brief overview of the typical pitfalls.
Explainability of AI
A major disadvantage of artificial intelligence, in particular machine learning based on neural networks, is the complexity of the algorithms. This makes them highly non-transparent, hence their designation of “black-box AI” (see Figure 2). The complex nature of AI algorithms not only concerns their mathematical description but also—in the case of deep-learning algorithms—their high level of dimensionality and abstraction. For these classes of AI, the extent to which input information contributes to a specific decision is mostly impossible to determine. This is why AI is often referred to as “black box AI”. Can we trust the prediction of the AI system in such a case and, in a worst-case scenario, can we identify a failure of the system or a misdiagnosis?
Figure 2. Implementation of artificial intelligence (AI) as black box. Given the non-transparency of the artificial intelligence system, it is impossible to say whether the result is correct.
A world-famous example of the result of a “black-box AI” was the match between AlphaGo, the artificial intelligence system made by DeepMind (Google) and the Go world champion, Lee Sedol. In the match, which was watched by an audience of 60 million including experts, move 37 showed the significance of these particular artificial intelligence characteristics. The experts described the move as a “mistake”, predicting that AlphaGo would lose the match since in their opinion the move made no sense at all. In fact, they went even further and said, “It’s not a human move. I’ve never seen a human play this move”.
None of them understood the level of creativity behind AlphaGo’s move, which proved to be critical for winning the match. While understanding the decision made by the artificial intelligence system would certainly not change the outcome of the match, it still shows the significance of the explainability and transparency of artificial intelligence, particularly in the medical field. AlphaGo could also have been “wrong”!
One example of AI with an intended medical use was the application of artificial intelligence for determining a patient’s risk of pneumonia. This example shows the risk of black-box AI in the MedTech sector. The system in question surprisingly identified the high-risk patients as non-significant.10 Rich Caruana, one of the leading AI experts at Microsoft, who was also one of the developers of the system, advised against the use of the artificial intelligence he had developed: “I said no. I said we don’t understand what it does inside. I said I was afraid.”11
In this context, it is important to note that “open” or “explainable” artificial intelligence, also referred to as “white box”, is by no means inferior to black-box AI. While there have been no standard methods for “opening” the black box, there are promising approaches for ensuring the plausibility of the predictions made by AI models. Some approaches try to achieve explainability based on individual predictions on input data. Others, by contrast, try to limit the range of input pixels that impact the decisions of artificial intelligence.
Right to Explainability
Medical devices and their manufacturers must comply with further regulatory requirements in addition to the Medical Device Regulation (MDR) and the In-vitro Diagnostics Regulation (IVDR). The EU’s General Data Protection Regulation (GDPR), for instance, is of particular relevance for the explainability of artificial intelligence. It describes the rules that apply to the processing of personal data and is aimed at ensuring their protection. Article 110 of the Medical Device Regulation (MDR) explicitly requires measures to be taken to protect personal data, referencing the predecessor of the General Data Protection Regulation.
AI systems which influence decisions that might concern an individual person must comply with the requirements of Articles 13, 22 and 35 of the GDPR.
“Where personal data … are collected…, the controller shall… provide….the following information: the existence of automated decision-making … and… at least in those cases, meaningful information of the logic involved…”
In simple terms this means, that patients who are affected by automated decision-making must be able to understand this decision and have the possibility to take legal action against it. However, this is precisely the type of understanding which is not possible in the case of black box AI. Is a medical product implemented as black-box AI eligible for certification as a medical device? The exact interpretation of the requirements specified in the General Data Protection Regulation is currently the subject of legal debate.
Verification and Validation of the AI model
The Medical Device Regulation places manufacturers under the obligation to ensure the safety of medical devices. Among other specifications, Annex I to the regulation includes, , requirements concerning the repeatability, reliability and performance of medical devices (both for stand-alone software and software embedded into a medical device):
“Devices that incorporate electronic programmable systems, including software, … shall be designed to ensure repeatability, reliability and performance in line with their intended use.“ (MDR Annex I, 17.1)15
Compliance with general safety and performance requirements can be demonstrated by utilizing harmonized standards. Adherence to a harmonized standard leads to the assumption of conformity, whereby the requirements of the regulation are deemed to be fulfilled. Manufacturers can thus validate artificial intelligence models in accordance with the ISO 13485:2016 standard, which, among other requirements, describes the processes for the validation of design and development in clause 7.3.7.
For machine learning two independent sets of data must be considered. In the first step, one set of data is needed to train the AI model. Subsequently, another set of data is necessary to validate the model. Validation of the model should use independent data and can also be performed by cross-validation in the meaning of the combined use of both data sets. However, it must be noted that AI models can only be validated using an independent data set. Now, which ratio is recommended for the two sets of data? This is not an easy question to answer without more detailed information about the characteristics of the AI model. A look at the published literature (state of the art) recommends a ratio of approx. 80% training data to approx. 20% validation data. However, the ratio being used depends on many factors and is not set in stone. The notified bodies will continue to monitor the state of the art in this area and, within the scope of conformity assessment, also request the reasons underlying the ratio used.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

02 Apr
Securing Medical Devices: What is Really Needed?
Alan Grau, VP of IoT/Embedded Solutions at Sectigo, wrote in MedTechIntelligence: “Medical device manufacturers continue to develop new, highly sophisticated and increasingly connected products. These products offer a wide range of benefits: Improved treatments, more precise diagnostics, better patient monitoring, automated control, and central reporting and monitoring of data.
This increased connectivity, however, comes with risks for both patients and providers.
Even though these companies are heavily investing in the development of new medical device technologies, they often lack the security expertise and resources to ensure high levels of security are built into these products. Many devices employ new protocols, platforms and middleware solutions that have not been thoroughly vetted for security issues. The result, not surprisingly, is a slew of devices that are easily compromised by hackers.
We continue to see headlines of new security vulnerabilities found in critical medical devices. Ransomware attacks have targeted hospitals and medical providers with alarming success. In these kinds of attacks, hackers compromise a system, encrypting critical data so the systems cannot operate, then require a ransom payment to restore the system to working order.
Past ransomware attacks have targeted IT and database systems, but future attacks may focus on medical devices. In fact, in 2019, cybersecurity experts found dangerous vulnerabilities in the technical design of some insulin pumps, prompting the FDA to issue a warning that hackers could compromise insulin pumps by connecting to them via WiFi and changing the pump’s settings to either under- or over-deliver insulin. If hackers are able to control systems that impact patient outcomes, they will have tremendous leverage for their ransom demands.
Use of multiple layers of protection, including firewalls, authentication, security protocols and intrusion detection/intrusion prevention, is a long-established driving principle for enterprise security. In contrast, most medical devices lack basic firewalls or security protocols, and often rely on little more than simple password authentication. There was an assumption that these devices are not attractive targets to hackers, or are not vulnerable to attacks, but that is no longer valid. Attacks against all types of embedded devices, including medical products, are on the rise, and greater security measures are now needed.
For more than 25 years, cybersecurity has been a critical focus for large enterprises. Now medical device design engineers need to take a page from the enterprise security playbook.
Embedded Security Challenges
Medical devices are very different from standard PCs. They are fixed-function devices designed to perform a specialized task. Installing new software on the system in the field often requires a specialized upgrade process or is simply not supported. These devices are optimized to minimize processing cycles and memory usage, so they lack extra processing resources. PC security solutions won’t solve the security challenges of these devices.
Challenges for medical device security include:
- Critical functionality: Medical devices control life-enabling systems and manage sensitive data.
- Replication: Once designed and built, medical devices are mass produced, resulting in thousands to millions of identical devices. Once discovered, a successful attack against one of these devices can be replicated across all the devices.
- Security assumptions: Many medical device engineers have long assumed that their products are not targets for hackers and have not considered security a critical priority.
- Patching limitations: Most medical devices are not easily upgraded. Once they are deployed, they will only run the software that was originally installed at the factory.
- Long lifecycle: The lifecycle for medical devices may be as long as 10, 15 or even 20 years. Building a device that will stand up to the security requirements of the next two decades is a tremendous challenge.
- Deployed outside of the enterprise security perimeter: Medical devices may be mobile or may be deployed in the home, environments lacking the protections found in a corporate environment.
- Siloed: No way for the end user to easily monitor the device’s security health or make changes.
Cyberattacks and The Motivated Hacker
The level of security required for a medical device varies depending upon the function of the device. Rather than asking if the device is secure, OEMs should be asking if the device is secure enough. A robotic surgical system clearly needs a very different level of security than sensors equipped with communication capability for remote monitoring of patients.
Hacking is not just the domain of bored teenagers, hacking drones, or even the small groups of motivated hackers. When the stakes are high enough, cyberattacks are typically multi-phased, multi-year efforts that are carried out by large, well-funded teams of hackers.
We are no longer talking about protecting a device from just malformed IP packets or DoS packet floods. Hackers often have detailed information about the device they are targeting and have sophisticated toolkits and skills that can be used to develop attacks.
Have you considered how to protect devices from attack by a group with detailed knowledge of the inner workings of your product?
FDA Cybersecurity Guidelines
The FDA has issued guidance for OEMs building medical devices. A few of the capabilities recommended in the FDA guidelines include:
- Restricting unauthorized access to medical devices
- Making certain that firewalls are up-to-date
- Monitoring network activity for unauthorized use
- Disabling all unnecessary ports and services
As an engineer working on a medical device, what do these guidelines mean? Many medical devices are specialized products and the security solutions used for standard PCs often won’t work for specialized devices. Clearly, meeting the security guidelines is important. Doing so requires an approach that is customized to the needs of the device.
Security Requirements for Medical Devices
A security solution for medical devices must protect firmware from tampering, secure the data stored by the device, secure communication, and protect the device from cyberattacks. This can only be achieved by building in security from the early stages of design.
There is no “one-size-fits-all security solution” for medical devices. Engineers must take into consideration the cost of a security failure (economic, environmental, social, etc.), the risk of attack, available attack vectors, and the cost of implementing a security solution. Table I outlines the features that need to be considered.
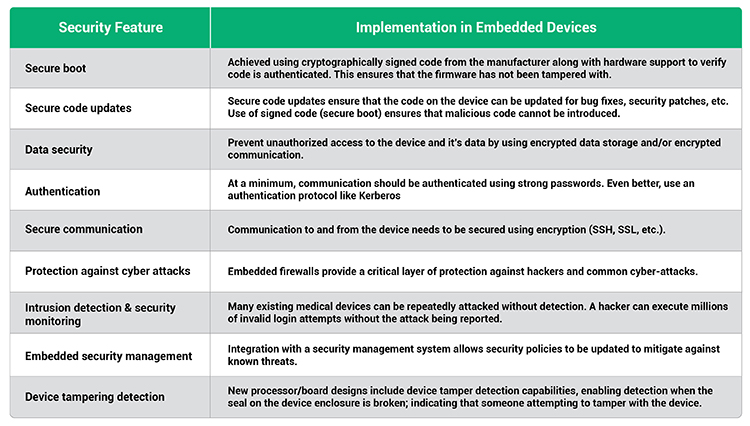
Table I. Security features that should be considered in embedded medical devices.
Integrating Security into The Device
Building protection into the device itself provides a critical security layer that ensures devices are no longer depending on the corporate firewall as their sole layer of security and allows security to be customized to the needs of the device. The engineering team must be as focused on security as they are on the new capabilities of the device. Building security capabilities into a medical device will enable the device to meet the FDA security guidelines.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

26 Oct
FDA – Digital Health Software Precertification (Pre-Cert) Program
FDA gives an update on the Software Precertification Pilot Program:
“What Is the Software Pre-Cert Pilot Program?
The Software Precertification (Pre-Cert) Pilot Program, as outlined in the FDA’s Digital Health Innovation Action Plan [PDF], will help inform the development of a future regulatory model that will provide more streamlined and efficient regulatory oversight of software-based medical devices developed by manufacturers who have demonstrated a robust culture of quality and organizational excellence, and who are committed to monitoring real-world performance of their products once they reach the U.S. market.
This proposed approach aims to look first at the software developer or digital health technology developer, rather than primarily at the product, which is what we currently do for traditional medical devices. Because software products can be adapted to respond to glitches, adverse events, and other safety concerns quickly, the FDA is working to establish a regulatory framework that is equally responsive when issues arise to help ensure consumers continue to have access to safe and effective products. In the Pre-Cert program, the FDA is proposing that software products from precertified companies would continue to meet the same safety and effectiveness standard that the agency expects for products that have followed the traditional path to market.
This pilot is an important first step to help us explore and evaluate the program model to inform how we establish the Pre-Cert Program. Pre-Cert 1.0, the first version of the program that was available for pilot testing within the FDA’s current authorities in 2019, is limited to manufacturers of software as a medical device (SaMD). Learn more about software as a medical device (SaMD)External Link Disclaimer.
As the FDA leverages insights from testing of Pre-Cert 1.0, we may find that the voluntary program could include software in a medical device (SiMD) or other software that could be considered accessories to hardware medical devices.
How Does the Pre-Cert Pilot Program Work?
The Software Precertification Pilot Program version 1.0 Working Model (PDF) explains how the FDA has reimagined its way of regulating digital health products and details the proposed key components of the Pre-Cert pilot program.
In 2019, the Software Pre-Cert program entered its test phase and the FDA shared the Test Plan (PDF).
The FDA’s Test Plan is designed to assess whether the Excellence Appraisal and Streamlined Review components together produce an equivalent basis for determining reasonable assurance of safety and effectiveness for a software device prior to its introduction to the market, as compared to the FDA’s traditional models.
The FDA describes how it plans to implement the Pre-Cert Pilot Program within its current authorities in the Regulatory Framework. Learn more about the Regulatory Framework for the Pre-Cert model.
In addition, the 2020 Update summarizes the FDA’s test activities, provides an update on our progress, shares what we have learned, and outlines next steps. For more information, see Developing the Software Precertification Program: Summary of Learnings and Ongoing Activities: 2020 Update [PDF – 1.5 MB].
Proposed Key Components of a Future Pre-Cert Program:
The FDA’s Total Product Lifecycle (TPLC) approach enables the evaluation and monitoring of a software product from its premarket development to postmarket performance, along with continued demonstration of the organization’s excellence. Learn more about this approach in the version 1.0 working model (PDF).
- Excellence Appraisal: Identifying the objective criteria and methodology that the FDA will use to pre-certify a company and decide whether a company can keep its precertification status.
The FDA is currently basing the Pilot Program’s criteria on five excellence principles: patient safety, product quality, clinical responsibility, cybersecurity responsibility, and proactive culture. The FDA is currently considering two levels of precertification based on how a company meets the excellence principles and whether it has demonstrated a track record in delivering safe and effective software products. - Review Determination: Developing a risk-based framework so a precertified company can determine the premarket review pathway for their products. Potentially precertified companies could market their lower-risk devices without the FDA’s premarket review or only a streamlined premarket review based on the company’s precertification level and International Medical Device Regulators Forum (IMDRF) risk categorization.
The FDA is planning to leverage the IMDRF framework, to the extent consistent with current statutory authority, to help determine the risk categorization of a SaMD product, incorporating information about the medical purpose of the SaMD and the seriousness of the medical condition that the SaMD is intended to treat.
The FDA is also considering appropriate means to educate patients and providers about the premarket review and postmarket monitoring obligations for each SaMD risk category. - Streamlined Review: Identifying the type of information that a precertified company would include in its premarket submission for the FDA to review software products for safety and effectiveness before patients access them.
The FDA is exploring using an interactive streamlined review of a SaMD with information the agency already has gained from the process to precertify a company, and additional information the company would share about the SaMD’s product performance, clinical association between the SaMD output and a clinical condition, and safety measures. - Real-world Performance: Identifying the type of information that may be available to or accessibly by a precertified company about how its software product is performing with patients to support the regulatory status of the product and new and evolving product functions.
The FDA is considering how best to work with a company to collect and interpret real-world information about a SaMD and to evolve the product’s safety and effectiveness to address any emerging risks. The sources of real world performance data may include information about a user’s experience, software performance data, and clinical outcomes.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

02 Oct
MedSysCon Medizintechnik GmbH

By loading the video, you accept YouTube’s privacy policy.
Learn more