
29 Sep
FDA launches Digital Health Center of Excellence
The FDA has launched its Digital Health Center of Excellence (DHCoE), as the agency continues with its commitment to advancing use of technology such as mobile health devices, software and wearables to create medical products.
The FDA announced: “The launch of the Digital Health Center of Excellence is an important step in furthering the agency’s overarching dedication to the advancement of digital health technology, including mobile health devices, Software as a Medical Device (SaMD), wearables when used as a medical device, and technologies used to study medical products.
“Establishing the Digital Health Center of Excellence is part of the FDA’s work to ensure that the most cutting-edge digital health technologies are rapidly developed and reviewed in the U.S.,” said FDA Commissioner Stephen M. Hahn, M.D. “Today’s announcement marks the next stage in applying a comprehensive approach to digital health technology to realize its full potential to empower consumers to make better-informed decisions about their own health and provide new options for facilitating prevention, early diagnosis of life-threatening diseases, and management of chronic conditions outside of traditional care settings. The Digital Health Center of Excellence will provide centralized expertise and serve as a resource for digital health technologies and policy for digital health innovators, the public, and FDA staff.”
The FDA will continue to build and formalize the coordinating structure and operation of the Digital Health Center of Excellence as part of an effort to modernize digital health policies and regulatory approaches, and provide efficient access to highly specialized expertise, knowledge, and tools to accelerate access to safe and effective digital health technology. The agency is appointing Bakul Patel as the first director. Bakul Patel has been leading regulatory and scientific efforts related to digital health devices at the FDA since 2010.
“The establishment of the Digital Health Center of Excellence is part of the planned evolution of the FDA’s digital health program to amplify the digital health work that is already being done and building upon years of work at the agency,” said Jeff Shuren, M.D., J.D., director of CDRH. “In the last several years, we have established partnerships internally and externally to coordinate digital health activities and to promote the consistency of regulatory policy while continuing to innovate in our regulatory approaches.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

05 Aug
Is Computer Software Assurance (CSA) the new CSV?
Bart Degroote, Senior Project Manager at pi life sciences consultancy, commented the FDA draft guidance on Computer Software Assurance within the PI knowledge page:
“The FDA draft guidance on Computer Software Assurance is a paradigm shift from document focused computer system validation to critical thinking assurance practices. The Guidance is on FDA’s list for release in September 2020 and applies to non-product quality system software solutions. The principles and approaches will apply to all regulated organizations.
How it all started
The concept of validation was derived from engineering validation principles that have been extended to the software industry. Feeling the necessity to validate computer systems, the FDA published the ‘bluebook’ (1983) this was a guide to the inspection of computerized systems in pharmaceutical processing.
These regulations on the use of electronic records and electronic signatures are formalized in 1997 in 21 CFR part 11 the FDA and revised in 2003, for Eudralex (EU GMP regulations) this is Annex 11 (EME 1998).
According to both computer system validation is defined as: “Confirmation by examination and provision of objective evidence that software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled”
Purpose and initial need for CSV
The purpose of the validation process is to provide a high degree of assurance that a computer system will consistently produce electronic records which meets predetermined specifications and quality attributes.
FDA regulations mandate the need to perform Computer System Validation and these regulations have the impact of the law. Having the evidence that computer systems are serving their intended purpose and operating properly represents a good business practice.
Failure to take corrective action immediately can result in shutting down manufacturing facilities, consent decrees, and stiff financial penalties.
Why we need a new approach
The Guidance on computer system validation is already dating back to 2003 in a rapidly changing digital landscape and is no longer aligned with new technologies. This makes it unclear on how much testing is enough and where to focus that testing.
FDA believes the use of automation, information technology and data solutions throughout the life cycle can provide significant benefits to enhance quality and patient safety. This substantial benefit in enhancing quality is shown in other industries utilizing thorough automation.
At this moment CSV is an obstacle in the process to move to a more automated environment, this due to the required extensive testing and comprehensive documentation that demonstrate the validation of computer software.
To support the use of this new technologies FDA is drafting new guidelines “Computer Software Assurance for Manufacturing, Operations, and Quality System Software”, CSA will tackle the issues we have at this moment with CSV.
Additionally, FDA intends to focus on direct impact systems and not on the indirect systems (support tools).
The paradigm shift of CSA
The new paradigm will focus on critical thinking (risk-based), assurance needs, testing activities and documentation in that order.
Risk determination should focus on:
- Does the software impact patient safety?
- Does this software impact product quality?
- How does this software impact your quality system integrity?
Comparison CSV VS CSA
Focus on creating documentary records for compliance VS Focus on testing for higher confidence in system performance
Validating everything with the risk to miss higher risk functionality VS Risk-based assurance, applying the right level of risk to patient safety and/or product quality
Ignoring previous assurance activity or related risk controls VS Taking prior assurance activities into account and upstream / downstream risk controls”
Please find the complete article here.
Please get in touch with us:
+49-176-57694801

24 Jul
DevOps – A Culture of Shared Responsibility
BCG recently published a fascinating insight into one of latest software industry best practices:
DevOps Takes Agile Further—and Across the Software Life Cycle
Thriving in the digital age requires levels of efficiency, agility, and quality in software development that can be delivered only by fully embracing DevOps practices, which combine development and operations in IT.
Agile methodologies have afforded many companies a much-needed tool in the digital era: a collaborative and flexible way to develop software. Cross-functional teams, quick and frequent iterations, and continual testing and feedback are powerful agile practices that have enabled companies to improve their development process. But businesses can realize even greater transformative change—and greater success—by implementing a second set of practices known as DevOps.
What Is DevOps?
DevOps combines the development and operations activities of an IT organization to increase collaboration and automation along the entire product lifecycle.
Specifically, the approach breaks down the walls within IT to embed employees from IT operations onto cross-functional agile teams working on software development. New processes and tools are used to align the IT and software development work.
This set of practices is not an alternative to agile but a complement. A DevOps approach takes agile a step further and applies it beyond the plan, design, build, and test stages of software development to the rest of the software lifecycle: deployment, release, operation, and monitoring.
The efficiency and speed gains that DevOps can deliver are particularly critical in today’s environment, when customers as well as technological developments are demanding ever-shorter development cycles and ever-increasing quality.
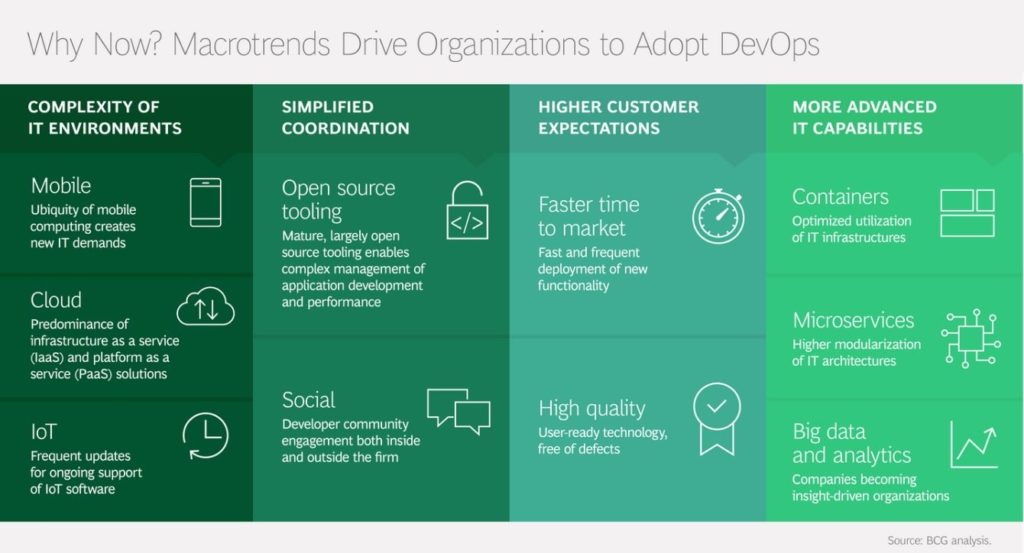
The Elements and Application of DevOps
In combination with agile, DevOps delivers six key elements that serve as the building blocks of a holistic transformation of software development. …”
Please find the complete article here.
Please get in touch with us for further support:
+49-176-57694801

19 Jun
FDA’s Transition From Computer System Validation To Computer Software Assurance
Kathleen Warner, Ph.D., RCM Technologies, reports in the recent issue of meddeviceonline.com about FDA`s transition from Computer System Validation to Computer Software Assurance:
“The FDA regulation 21 CFR Part 11 in 1997 and the related guidance of 2003 paved the road to implementation of computer system validation (CSV) by life sciences companies.
As pharmaceutical companies perfected their business processes and became more efficient in validating computer systems, the piles of documentation continued to grow without significant quality benefits. The focus was on speed, documentation accuracy and completeness, inspections, audits, and complying with the regulation.
In 2011 the Center for Devices and Radiological Health (CDRH) initiated the Case for Quality, a new program that identified barriers in the current validation of software in medical devices guidance (released in 2002). Now, CDRH — in cooperation with the Center for Biologics Evaluation and Research (CBER) and the Center for Drug Evaluation and Research (CDER) — is preparing to release new guidance, Computer Software Assurance for Manufacturing, Operations and Quality Systems Software, in late 2020.
This new guidance will provide guidelines for streamlining documentation with an emphasis on critical thinking, risk management, patient and product safety, data integrity, and quality assurance. Even though this guidance is being developed for the medical device industry, the FDA has indicated it should be considered when deploying non-product, manufacturing, operations, and quality system software solutions such as quality management systems (QMS), enterprise resource planning (ERP) systems, laboratory information management systems (LIMS), learning management systems (LMS), and electronic document management systems (eDMS). As such, the guidance will be applicable to research and development (R&D), clinical, laboratory, and other groups within pharmaceutical, biopharmaceutical, and medical device companies that are currently meeting the regulations for electronic records and electronic signatures (ERES)1 and computer system validation (CSV).2
As the digital world evolves, new technologies, including automation and artificial intelligence (AI), are hitting the marketplace and focusing on quality through unscripted and automated testing and smart applications. This paper describes how computer system validation (CSV) is performed today, introduces the concept of computer software assurance (CSA), and discusses how CSA can refocus validation on what’s important in the new world of digital technologies.
Computer System Validation is the process of achieving and maintaining compliance with the relevant GxP regulations defined by the predicate rule.3 Fitness for intended use is achieved by adopting principles, approaches, and life cycle activities. The validation methodology determines the framework to follow in developing the validation plans and reports and applying appropriate operational controls throughout the life cycle of the system.
CSV is typically performed following the standard operating procedures (SOPs) for the software development life cycle (SDLC) and the CSV, respectively. According to GAMP 5, the computerized system validation methodology comprises four distinct life cycle phases: Concept, Project, Operation, and Retirement.
- Concept Phase: During this phase, activities are initiated to justify project commencement.
- Project Phase: During this phase, the overall system is implemented and tested according to preapproved documents. The SDLC includes, but is not limited, to the following: validation methodology and plan, requirements, software documentation and test plans, traceability matrix, and summary reports.
- Operation Phase. During this phase, routine, day-to-day activities associated with the system (as defined in user manuals, SOPs, work instructions, etc.) are performed and include, but are not limited to, the following: backup and restore, disaster recovery, change management, incident/deviation management, access and security management, and periodic review.
- Retirement Phase. During this phase, the system is no longer used, needed, or operational and is at the end of its life cycle. A retirement/decommissioning plan is developed to document the approach and tasks to manage the data and records.”
Please find the complete article here.
Please get in touch with us:
+49-176-57694801

22 May
MDCG 2019-16 – Guidance on cybersecurity for medical devices
MD101 comments in their recent blog the new guidance on cybersecurity for medical devices: the MDCG 2019-16: “This guidance covers a broad range of topics applicable to all stakeholders in the medical device supply chains, and to end-users. It explains a bit why it is 46 pages long.
Section 1 – Introduction
The first section of the guidance draws the link between regulatory requirements and cybersecurity processes / artifacts. The figures 1 & 2 are quite a good synthesis of the MDR requirements covering cybersecurity. Note that these figures could be duplicated with several other topics, like electrical security, biocompatibility (and so on), and state-of-the-art applicable standards.
The topics listed in section 1.4 cover all topics where cybersecurity is involved. This list is very useful to assess where cybersecurity requirements shall be implemented in your QMS processes. E.g: design controls, post-market surveillance.
Section 2 – Basic cyber concepts
If you’ve already read documents like AAMI TIR 57 or the 2 FDA guidances on cybersecurity, you will retrieve in this section some information common to these documents. The novelty in this MDCG guidance is the link between these concepts and the MDR General Safety and Performance Requirements (GSPR).
We also retrieve in this section concepts of defense in depth, good security hygiene (basic security hygiene in FDA guidance), and the relationship between security risks and safety risks.
Another concept is introduced, not so easily found in other guidance: the link between usability engineering and cybersecurity:
[a vulnerability] should be regarded as an enabler of reasonably foreseeable misuse .
If an exploit exists, an user will use it with a probability to assess, linked to the user’s education level and the ease of exploitation based on use scenarios.
The concept of joint responsibility is also introduced. All stakeholders in the supply chain shall take their part of the job: medical device integrators operators, and users. Manufacturers, don’t be fooled by this joint responsibility: as usual, your responsibility will be engaged in case of adverse event. You shall have established processes to ensure the proper installation, deployment, configuration, and exploitation of your devices in a secure manner. Simply said, this joint responsibility doesn’t exonerate manufacturers. Quite the opposite, it engages the responsibility of other stakeholders.

Section 3 – Secure design and manufacture
Section 3 delves into technical details (as far as a guidance can do, it’s not a standard), with a list of good practices to ensure that the device is secure by design. These 6 best practices can be seen as the steps of processes found in IEC 62304 design and maintenance processes:
Security management:
4.1 of ISO 13485, for the security risk management process
5.1 of IEC 62304: Software development plan
6.1 of IEC 62304: Software maintenance
Specification of Security Requirements:
5.2 of IEC 62304: software requirements analysis
Secure by design, including defense in depth:
5.3 of IEC 62304: software architecture
Secure implementation:
5.4 of IEC 62304: software detailed design
5.5 of IEC 62304: software implementation and unit verification
and a precision on SOUP management
Secure Verification and Validation
5.6 of IEC 62304: software integration testing
5.7 of IEC 62304: software system verification
Management of security-related issues
6.2 of IEC 62304: Problem and modification analysis
9 of IEC 62304: Problem resolution
Security update management
6.3 of IEC 62304: Modification implementation
8.2 of IEC 62304: Change control
Security guidelines:
5.8 Software release
and also software documentation, see IEC 82304-1 section 7.
Security Risk management
Section 3.2 continues with recommendations on the security risk management process, especially the link between security risks and their impact on safety. A very important remark is present in this section, for the sake of clarity of safety risk management reports: safety risk assessment might list generic security related hazards (…). This is to avoid detailing every possible attack vector.
This section also insists on the fact that compliance to GSPR 1 to 4 requires to assess security risks. Thus, cybersecurity isn’t nested only in GSPR 17.2 on software, but is promoted to the first main GSPR’s.
Security capabilities
Section 3.3 lists some possible security capabilities for software. This list is absolutely not exhaustive. This is a good starting point, though.
Interestingly, this section also contains an analogy between the precedence of safety mitigation actions (section 6.2 of ISO 14971) and their security risk equivalent. Worth reading!
This section ends with a remark on the need to maintain safety and effectiveness but also performance requirements and efficiency of mitigation actions, with security capabilities. New columns to your risk assessment matrices?”
Please find the complete article here.
Please get in touch with us:
+49-176-57694801

08 May
Cybersecurity Vulnerabilities in Healthcare
Ben Hartwig, Web Operations Executive, InfoTracer gives in a recent article of the Medical Product Outsourcing Magazine (MPO) an overview about the “2020 MedTech Cyberchaos”.
The issue with medical technology is the cybersecurity isn’t up to the level of where it needs to be, and criminals know this. Manufacturers that build these products have a treasure trove of sensitive information on vendors and clients alike. If medical device makers don’t perform their due diligence by conducting a criminal records search on new employees, one bad actor on the inside can steal all their valuable data or give network access to hackers from the outside.
Weak Links in the Medtech Manufacturing Industry
While some medical manufacturers are shoring up their cybersecurity response, others aren’t, and they may be exposing their weak IT infrastructure to cyberattacks. Following are the most vulnerable network points that need immediate attention.
Compromised Cloud Infrastructure
Plenty of medical manufacturers are leveraging the power of the cloud to advance healthcare technology because sharing information with speed and efficiency can save lives. However, the lack of cloud computing professionals working in these companies or the current staff’s lack of knowledge in port data security and legacy systems can lead to holes in the cloud architecture that cybercriminals can exploit. Regular scanning can detect these vulnerabilities, but not all manufacturers are up to the task.
Open-Source Software
While using open-source software isn’t inherently wrong, developers need to devote their time and effort to make proprietary changes to the code that improves security and locks it down so no one outside the organization can penetrate it. By definition, open-source software means everyone has the same access to the lines of code that make it up. If a lazy developer uses the code “as is” or installs an untested program from an unsecured hard drive, hackers can easily bypass and infiltrate the system.
Poor Internal Security Protocols
Some manufacturers don’t have stringent internal security policies in place to secure the premises from rogue employees and protect their sensitive data. As harsh as it sounds, the weakest link in any organization will always be the employees. This is why the vetting process before hiring should include criminal records search and other checks to ensure the person is legit even before he or she steps inside the building. There should be a culture of data protection instilled in everyone, so no one can make the common mistake of using unsecured freeware, using unsecured WiFi, or responding to phishing attempts. Employees should be informed about tools such as email lookup, and username search that can help them to be protected from common scams.
Please find the complete article here.
Please get in touch with us:
+49-176-57694801

02 May
Healthcare: Confidence in Cloud Computing grows
healthcare-in-europe.com comments on Johan Sjöberg, a medical physicist at Karolinska University Hospital in Stockholm, Sweden, who claims that cloud computing in healthcare is a couple of decades behind the rest of society.
“The purpose of a well-functioning healthcare system is to provide excellent care to its patients at the lowest cost possible. This is what value-based healthcare is all about,” says Sjöberg. Unfortunately, many healthcare providers aren’t entirely there yet. “What is a cloud? It’s just somebody else’s computer, right? There’s nothing magical about it,” Johan Sjöberg suggests. “But there are a lot of benefits that are attributed to the concept of a cloud service. “You have a central repository. It’s simple to build interfaces between the different layers in the databases. That offers the opportunity to roam the data and actually pull some interesting information from those databases, which is valuable to our patients. It’s a shared platform. It offers a lot of opportunities, not just for data management, but also for communication management.”
Sjöberg notes that the amount of data generated in healthcare today is “quadrupling every second year”, yet the challenge lies in figuring out if there’s anything interesting in that health data.
What the cloud environment does is break down the silo walls and enables a more holistic view on patient care, according to Thomas Friese, Head of Digital Platform for Siemens Healthineers. He notes that many leading-edge cloud solutions are forward looking in terms of the applications and benefits they can offer a healthcare enterprise. “The cloud offers a good opportunity to easily add on or migrate to new applications; it enables you to easily evolve.”
Continuously improving care
Cloud computing allows healthcare enterprises to utilize the latest technology at a fraction of the cost and deployment time of a local installation. For example, providers of cloud-based AI applications are highly scalable and can use a practically unlimited number of Graphical Processing Units (GPUs). A cloud environment seamlessly unites healthcare professionals in a large-scale team effort, making the knowledge and insights of individual professionals available across a global network. Big data becomes readily available, accessible, and easy to analyze as data sets located anywhere in the world are available to improve diagnostic capabilities, provide integrated decision support, and help physicians get a comprehensive view of a patient’s condition. Individualized treatment plans can increasingly be developed from valuable quantitative data. Enterprise-wide artificial intelligence-based assistants can serve as a clinical decision support mechanism (CDSM) for referring physicians ordering imaging tests.
Please find the complete article here.
Please get in touch with us for any further support:
+49-176-57694801

13 Apr
FDA issues Digital Health Innovation Action Plan
On March 26, 2020, the FDA published a paper called “Digital Health Innovation Action Plan” outlining the efforts to reimagine the FDA’s approach to ensuring all Americans have timely access to high-quality, safe, and effective digital health products. As part of this plan, the FDA committed to several key goals:
- Issuing guidance to modernize their policies.
- Increasing the number and expertise of digital health staff at the FDA.
- Developing the Digital Health Software Precertification Pilot Program (“Pre-Cert”).

From mobile medical apps and software that support the clinical decisions doctors make every day to artificial intelligence and machine learning, digital technology has been driving a revolution in health care. Digital health tools have the vast potential to improve our ability to accurately diagnose and treat disease and to enhance the delivery of health care for the individual. Digital tools are giving providers a more holistic view of patient health through access to data and giving patients more control over their health. Digital health offers real opportunities to improve medical outcomes and enhance efficiency.
How Is the FDA Advancing Digital Health?
The FDA’s Center for Devices and Radiological Health (CDRH) has established the Digital Health program, which seeks to better protect and promote public health and provide continued regulatory clarity by:
- Fostering collaborations and enhancing outreach to digital health customers, and
- Developing and implementing regulatory strategies and policies for digital health technologies.
How Are Digital Health Products Used?
Providers and other stakeholders are using digital health technologies in their efforts to:
- Reduce inefficiencies,
- Improve access,
- Reduce costs,
- Increase quality, and,
- Make medicine more personalized for patients.
Patients and consumers can use digital health technologies to better manage and track their health and wellness related activities.
The use of technologies, such as smart phones, social networks, and internet applications, is not only changing the way we communicate, but also providing innovative ways for us to monitor our health and well-being and giving us greater access to information. Together, these advancements are leading to a convergence of people, information, technology, and connectivity to improve health care and health outcomes.
Why Is the FDA Focusing on Digital Health?
Many medical devices now have the ability to connect to and communicate with other devices or systems. Devices that are already FDA approved, authorized, or cleared are being updated to add digital features. New types of devices that already have these capabilities are being explored.
Many stakeholders are involved in digital health activities, including patients, health care practitioners, researchers, traditional medical device industry firms, and firms new to the FDA regulatory requirements, such as mobile application developers.
The following are topics in the digital health field on which the FDA has been working to provide clarity using practical approaches that balance benefits and risks:
- Artificial Intelligence and Machine Learning (AI/ML) in Software as a Medical Device
- Cybersecurity
- Device Software Functions, including Mobile Medical Applications
- Health IT
- Medical Device Data Systems
- Medical Device Interoperability
- Software as a Medical Device (SaMD)
- Telemedicine
- Wireless Medical Devices
Please find the complete FDA article here.
Please get in touch with us for any further support:
+49-176-57694801

10 Apr
EU permits remote notified body audits during pandemic
Medtechdive.com reports that the European Commission’s Medical Device Coordination Group (MDCG) is allowing notified bodies to perform remote audits during the coronavirus outbreak, according to a guidance posted Wednesday. The MDCG detailed how notified bodies can run audits while quarantine orders and travel restrictions are stopping them from conducting site visits. The pandemic-oriented guidance the day after the Council of the European Union responded to the Commission’s proposed delay of the Medical Device Regulation. The Council’s planned amendments to the proposal clear up the confusion over whether the delay applies to all aspects of MDR.
Travel restrictions across the EU are stopping notified bodies from performing the on-site assessments central to their work. Recognizing that threat to the supply of medical devices, MDCG, an advisory group consisting of member state officials and industry players, laid out temporary measures notified bodies can take to work around the restrictions.
In some circumstances, notified bodies may perform remote audits. That option is open to notified bodies that have to carry out surveillance and recertification audits under the device directives, as well as when a manufacturer has filed a change notification or switched to a different notified body.
MDCG developed the guidance primarily to help notified bodies designated under the outgoing device directives. However, the advisory group said the flexibility provided in the guidance may apply to MDR and IVDR “in the event that the availability of devices is affected by COVID-19 restrictions.”
If an audit falls within the scope of the guidance, a notified body can leverage technology to perform a remote assessment. The guidance permits the off-site assessment of documents and records, and use of results from audits run under the Medical Device Single Audit Program, an initiative involving FDA and other regulators outside of the EU.
MDCG expects notified bodies to assess how to make use of the flexibilities provided by the guidance on a case-by-case basis. In doing so, notified bodies should consider the risks of each case, for example by factoring in their experience of the manufacturer and its compliance record.
The group is still developing guidance on the operational implementation of the principles.
Details of the flexibilities available to notified bodies emerged a day after the Council advanced the planned delay of the MDR date of application.
Please find the complete FDA article here.
Please get in touch with us for any further SaMD support:
+49-176-57694801

04 Apr
FDA proposed Regulatory Framework for Al/ML- Software as a Medical Device
On April 2, 2019, the FDA published a discussion paper “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) – Discussion Paper and Request for Feedback” that describes the FDA’s foundation for a potential approach to premarket review for artificial intelligence and machine learning-driven software modifications.

FDA proposed Regulatory Framework for Al/ML Software
Artificial intelligence and machine learning technologies have the potential to transform health care by deriving new and important insights from the vast amount of data generated during the delivery of health care every day. Medical device manufacturers are using these technologies to innovate their products to better assist health care providers and improve patient care. The FDA is considering a total product lifecycle-based regulatory framework for these technologies that would allow for modifications to be made from real-world learning and adaptation, while still ensuring that the safety and effectiveness of the software as a medical device is maintained.
What is Artificial Intelligence and Machine Learning?
Artificial Intelligence has been broadly defined as the science and engineering of making intelligent machines, especially intelligent computer programs (McCarthy, 2007). Artificial intelligence can use different techniques, including models based on statistical analysis of data, expert systems that primarily rely on if-then statements, and machine learning.
Machine Learning is an artificial intelligence technique that can be used to design and train software algorithms to learn from and act on data. Software developers can use machine learning to create an algorithm that is ‘locked’ so that its function does not change, or ‘adaptive’ so its behavior can change over time based on new data.
Some real-world examples of artificial intelligence and machine learning technologies include:
- An imaging system that uses algorithms to give diagnostic information for skin cancer in patients.
- A smart electrocardiogram (ECG) device that estimates the probability of a heart attack.
How are Artificial Intelligence and Machine Learning Transforming Medical Devices?
Adaptive artificial intelligence and machine learning technologies differ from other software as a medical device (SaMD) in that they have the potential to adapt and optimize device performance in real-time to continuously improve health care for patients. The International Medical Device Regulators Forum (IMDRF) defines software as a medical device as software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device. The FDA under the Federal Food, Drug, and Cosmetic Act (FD&C Act) considers medical purpose as those purposes that are intended to treat, diagnose, cure, mitigate, or prevent disease or other conditions.
How is the FDA Considering Regulation of Artificial Intelligence and Machine Learning Medical Devices?
The FDA predicts that under its current guidance, many changes made to software as a medical device driven by artificial intelligence and machine learning would be subject to a premarket review—this has prompted the FDA to reimagine a regulatory approach for these devices.
Traditionally, the FDA reviews medical devices through an appropriate premarket pathway, such as premarket clearance (510(k)), De Novo classification, or premarket approval. The FDA may also review and clear modifications to medical devices, including software as a medical device, depending on the significance or risk posed to patients of that modification. Learn the current FDA guidance for risk-based approach for 510(k) software modifications.
The FDA’s traditional paradigm of medical device regulation was not designed for adaptive artificial intelligence and machine learning technologies. Under the FDA’s current approach to software modifications, the FDA anticipates that many of these artificial intelligence and machine learning-driven software changes to a device may need a premarket review.
On April 2, 2019, the FDA published a discussion paper “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) – Discussion Paper and Request for Feedback” that describes the FDA’s foundation for a potential approach to premarket review for artificial intelligence and machine learning-driven software modifications.
The ideas described in the discussion paper leverage practices from our current premarket programs and rely on IMDRF’s risk categorization principles, the FDA’s benefit-risk framework, risk management principles described in the software modifications guidance, and the organization-based total product lifecycle approach (also envisioned in the Digital Health Software Precertification (Pre-Cert) Program).
In this framework, the FDA introduces a “predetermined change control plan” in premarket submissions. This plan would include the types of anticipated modifications—referred to as the “Software as a Medical Device Pre-Specifications”—and the associated methodology being used to implement those changes in a controlled manner that manages risks to patients —referred to as the “Algorithm Change Protocol.”
In this approach, the FDA would expect a commitment from manufacturers on transparency and real-world performance monitoring for artificial intelligence and machine learning-based software as a medical device, as well as periodic updates to the FDA on what changes were implemented as part of the approved pre-specifications and the algorithm change protocol.
The proposed regulatory framework could enable the FDA and manufacturers to evaluate and monitor a software product from its premarket development to postmarket performance. This potential framework allows for the FDA’s regulatory oversight to embrace the iterative improvement power of artificial intelligence and machine learning-based software as a medical device, while assuring patient safety.
Please find the complete FDA article here.
Please get in touch with us for any further SaMD support:
+49-176-57694801

25 Mar
EU plans to postpone the MDR by one year
According the EU spokesperson for public health and food safety, Stefan de Keersmaecker, the EU commission is working on a proposal to postpone the EU MDR by one year. This is mainly due to Covid-19 and should release the pressure on governments and manufacturers so they can focus on this crisis.
Please hear the statement of Stefan de Keersmaecker here.
The European Commission published the following statement:
“Commission working on proposal to postpone MDR application date for one year
Today (25 March 2020), the Commission announced that work on a proposal to postpone the date of application for the Medical Device Regulation (MDR) for one year is ongoing. The decision was reached with patient health and safety as a guiding principle.
The Commission is working to submit this proposal in early April for the Parliament and the Council to adopt it quickly as the date of application is the end of May. This decision will relieve pressure from national authorities, notified bodies, manufacturers and other actors and will allow them to fully focus on urgent priorities related to the coronavirus crisis.”
Please get in touch with us for any further MDR support:
+49-176-57694801
16 Mar
FDA okays Roche’s COVID-19 diagnostic test
PMLIVE reported in its pharma news that the US Food and Drug Administration (FDA) has approved Roche’s diagnostic test for the novel coronavirus, authorising it for emergency use. The cobas SARS-CoV-2 test is intended for qualitative detection of the novel coronavirus (SARS-CoV-2), which causes the respiratory disease COVID-19. Nasopharyngeal and oropharyngeal swab samples are taken from patients who have symptoms consistent with COVID-19, with these sample then sent to hospitals and laboratories for testing on Roche’s cobas 6800/8800 systems. These systems can produce test results in three and half hours, with a total of 1,440 results provided by the cobas 6800 system and 4,128 results by the cobas 8800 system within a 24-hour period. According to the Swiss pharma, millions of tests a month will be available for use on both systems, which it said offers improved operating efficiency and flexibility.
Please read the full article here
Please get in touch with us for any further support:
+49-176-57694801

12 Mar
FDA postpones foreign medical device inspections due to COVID-19 concerns
The U.S. Food and Drug Administration is postponing inspections outside of the U.S. in response to the COVID-19 outbreak. The FDA is postponing most foreign inspections through April 2020, effective immediately. Inspections outside the U.S. deemed mission-critical will still be considered on a case-by-case basis. For medical device manufacturers based outside the US which are dependent on FDA inspections to obtain US market registration, this announcement may mean delays in premarket application reviews.
FDA states that although temporarily not being able to physically inspect foreign produced FDA-regulated products or manufacturers, as an interim measure FDA will employ additional tools to ensure the safety of products imported to the U.S., which have proved effective in the past. These include denying entry of unsafe products into the U.S., physical examinations and/or product sampling at our borders, reviewing a firm’s previous compliance history, using information sharing from foreign governments as part of mutual recognition and confidentiality agreements and requesting records “in advance of or in lieu of” on-site drug inspections. For example, FDA began exercising this authority when postponing on-the-ground inspections of manufacturers of FDA-regulated products in China earlier in the outbreak. This is all part of the FDA’s multi-pronged and risk-based approach to ensuring quality, as well as compliance, with applicable federal laws and regulations.
As this remains a dynamic situation, FDA will continue to assess and calibrate their approach as needed to help advance federal response efforts in the fight against this outbreak.
Please read the full FDA statement here
Please get in touch with us for any further support:
+49-176-57694801

23 Feb
Major conglomerates bet on healthcare
Medtechdive.com reports about major global conglomerates like 3M and Philips have taken steps to increase focus on healthcare in recent years, shedding units covering other industries while buying additional medical assets in the belief the aging population will drive growth. GE took a different path, initially planning to spin off its healthcare unit (like Siemens did with a 2018 Healthineers IPO), only to decide to keep it in the fold. Across each company, healthcare is now a key element of growth forecasts, made clear during earnings reports this week.
Please read the full article here.
Please get in touch with us for any further support:
+49-176-57694801