
20 Jun
Clinical Decision Support Software Regulations
Ruth Beckers and Pascale Van Hoydonck wrote an groundbreaking article about the application of Clinical Decision Support Software regulations which was published by Kerman University of Medical Sciences: ”Impact of the regulatory framework on medical device software manufacturers: are the guidance documents supporting the practical implementation?; Comment on “Clinical decision support and new regulatory frameworks for medical devices: are we ready for it? – a viewpoint paper”. Int J Health Policy Manag. 2023;x(x):x–x. doi: 10.34172/ijhpm.2023.7470”
Abstract
The increasing use in clinical practice of software such as mobile apps and clinical decision support software has only recently been taken up by regulators around the world. Specifically, the European Commission and the US Food and Drug Administration have updated their regulatory framework in the last years. Van Laere S. et al. 1 have given an extensive overview of the European and US approach to regulate clinical decision support software. This commentary further discusses regulatory differences between the two geographies and their impact on manufacturers of medical device software. We discuss the practical implementation of the regulatory framework for medical device software (especially clinical decision support software) with a reference to the available international guidance documents and their limitations. Given the direction of stricter regulatory oversight in Europe, additional European guidelines/examples are desirable to enable a pragmatic regulatory approach ensuring continued access to innovative medical device software for European patients.
Keywords: Clinical Decision Support Software; Practical Implementation; Regulatory Framework; International Guidance Documents
Introduction
Just as other sectors, the healthcare sector has recently experienced an increased digitalisation. This tendency caused the widespread use of software, for example to streamline administrative processes within hospitals and care centres (IT equipment), but also to support clinical decision making by healthcare professionals. Van Laere S. et al.1 explain the different regulatory frameworks that apply to clinical decision support software in the United States(Food and Drug Administration, FDA, 21st Century Cures Act)2 and in Europe (EU Medical Device Regulation (MDR)3.
We will mainly comment and ask critical questions on the practical implementation of the regulatory framework for medical device software (especially clinical decision support software), with a reference to the available international guidance documents and their limitations. As mentioned by Van Laere S. et al.1, manufacturers of medical device software need to consider those regulatory frameworks carefully as they might impact their time and costs to market significantly.
Importance of guidelines keeping up to date with the “state of the art” software
There is a general trend of digitalization in healthcare with a broad spectrum of different medical and non-medical device software functions used. This is confirmed by policy makers through providing illustrative examples in guidance documents for the qualification of software used in the healthcare environment4-10. It is understood that the examples in the guidelines have been drafted in the light of today’s state of the art. However, we hope that those guidance documents will be treated by policy makers as living documents that will be updated with innovative examples (e.g., software algorithms that operate via machine learning or other artificial intelligence techniques) following the pace of evolving technologies. It is correctly noted in the viewpoint article that there is an assumption that MDR will hamper software development as the FDA Guidances 4-9 present more innovative software examples than the EU Medical Device Coordination Group (MDCG) Guidance Document10.
Qualifying a software product as a medical device – is the definition clear?
To be qualified as a medical device, a product must first fulfil the definition of a medical device according to the applicable jurisdiction. The international harmonization body (former Global Harmonization Task force (GHTF) or International Medical Device Regulators Forum (IMDRF) has created non-binding guidance documents (e.g. definition of the term “Medical Device”) to encourage regulatory systems’ convergence at the global level by eliminating differences between jurisdictions. This harmonized guidance would ideally result in decreasing the cost of gaining regulatory compliance and in allowing patients earlier access to innovative technologies and treatments11. The EU MDR definition and its specification of the medical purpose of a device is more leaning towards the GHTF definition compared to the device definition in section 201 (h) of the Food, Drug & Cosmetic Act.2 In addition, the term “monitoring” in the EU MDR3 is focused on products intended to monitor physiological processes, while “monitoring” is not present in the FDA device definition2. Unfortunately, there is no clear definition of the term “to monitor” which complicates the qualification of a software product. We would suggest defining monitoring as “following the evolution of a disease, injury/disability or physiological or pathological process or state at different stages or at different moments in time”. Many software products are intended to follow up chronic patients at home by visualising parameters measured with different hardware medical devices and notifying deviations to enable healthcare professionals to take treatment decisions. For such software products, we would welcome a clear definition of medical device monitoring in updated guidance documents to facilitate their qualification.
As confirmed by the FDASIA report12, the development of software products used to take decisions with diagnosis or therapeutic purposes has increased in the last decennia (called “decision support software in EU”10 or “clinical decision support (CDS) software in US”5). Unfortunately, no harmonized definition is available for CDS software. The authors of the viewpoint article came up with their own definition on CDS software: ‘any software system that integrates personal patient data with external sources of medical knowledge to assist the healthcare professionals in their decision-making process’1. Following MDCG guidance10, decision support software is intended to provide healthcare professionals and/or users with recommendations for diagnosis, prognosis, monitoring and treatment of individual patients. Following FDA guidance5, CDS software would provide ‘health care professionals (HCP) and patients with knowledge and person-specific information, intelligently filtered, or presented at appropriate times, to enhance health and health care. Generally, the meaning of those definitions seems identical, however limitations exist in the practical qualification of CDS software as a medical device. Van Laere S. et al.1 are correctly requesting further guidance on how FDA will assess the availability of plain language description of the logic or rationale used by an algorithm and the availability of the elements forming the basis of the recommendations to the intended user. The understanding of the basis of a certain recommendation might trigger that CDS software being exempted from the 21st Century Cures Act2. This approach of a software recommendation for being or not being understandable has not been considered in the EU MDCG guidance document as a criterium to qualify CDS software as a medical device. Additional clarification in guidance documents (US and EU) on the basis of the software recommendation and/or action of the software on the data (e.g. data analysis) would improve the qualification assessment of software products.
Is the classification of clinical decision support software different between jurisdictions?
The EU and FDA medical device legislations have leveraged the IMDRF risk-based framework for categorizing Software as a Medical Device (SaMD), based on the risk to patients if the software malfunctions.13 IMDRF’s categorization criteria and framework (Table 1)13 are not a regulatory classification, nor imply a convergence of classifications rules. Each jurisdiction has aligned her SaMD or medical device software (EU term) classification rules with this IMDRF framework in a unique way. As indicated by Van Laere et al., the EU MDR classification is much more stringent, especially due to the new classification Rule 11. Software intended to provide information in the clinical management is always classified as a Class IIa medical device in Europe, requiring the regulatory approval by a notified body (Table 2).3,10
The FDA approach is more pragmatic. FDA has drafted guidance on CDS software functions with specific focus on CDS software used for informing clinical management5. The categorization method for regulatory oversight is based on the user (a HCP, health care professional, or a patient or caregiver) on the one hand and on the fact that the user can review the basis of the information on the other hand (Table 3). With this approach, FDA strikes the right balance between ensuring patient safety and promoting innovation by clarifying which products would be the focus of FDA’s oversight and which would not. As noted by Van Laere S. et al.1, two medical devices with the same intended use may obtain different classifications in those two jurisdictions. Unfortunately, EU MDR does not consider the lower significance of informing clinical management of the IMDRF framework nor the stage of healthcare condition of the patient, to allow a lower classification and thus regulatory burden for such clinical decision support software (leading to a class IIa classification for category I.i in Table 2).
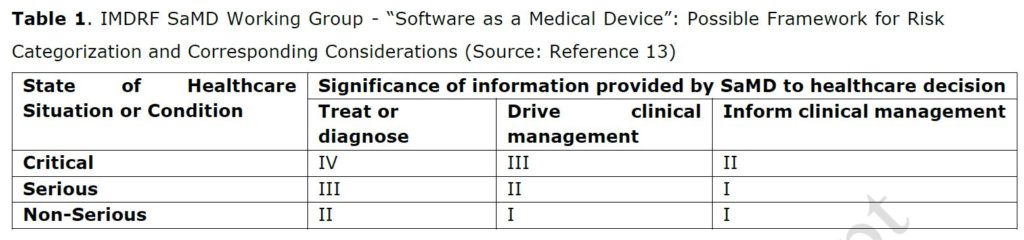
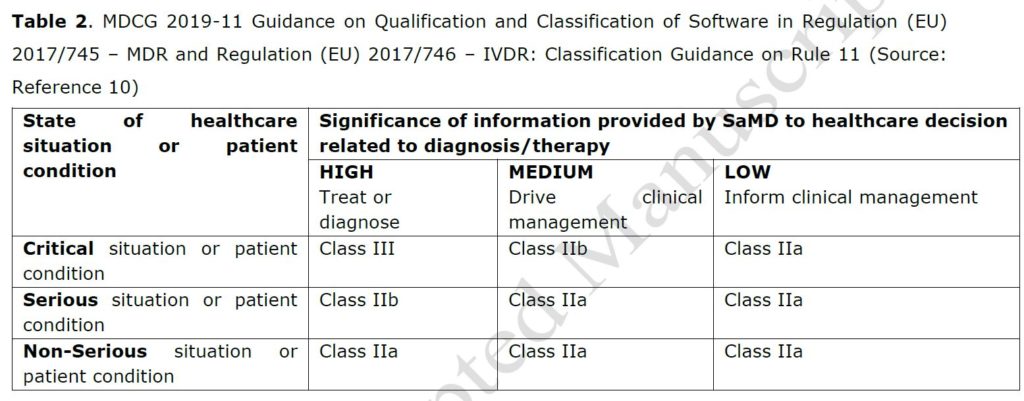
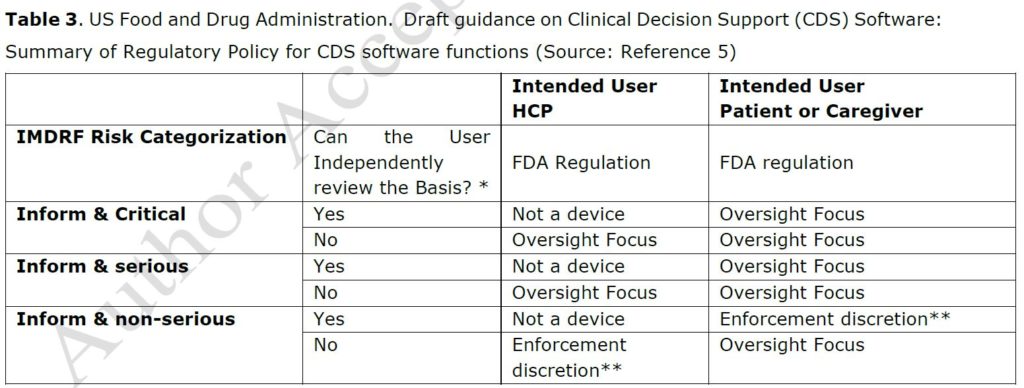
* “Can the User Independently Review the Basis?” asks whether the function is intended for the purpose of enabling the user to independently review the basis for the recommendations so that it is not the intent that user relies primarily on any such recommendation
** “Enforcement Discretion” indicates that, based on our current understanding of the risks of these devices, does not intend at this time to enforce compliance with applicable device requirements.
Are there other limitations in the practical implementation of medical device legislations?
Advanced technologies such as artificial intelligence (AI) are becoming more frequently used in healthcare for diagnostic or therapeutic purposes. The US FDA clearly has taken the path forward for embracing this technology by drafting an Artificial Intelligence/Machine Learning software as a medical device action plan14. In Europe, guidance on interpretation of significant changes to AI are not yet available. Due to the promotion of those innovative products to the market, there is a growing need for software experts in Europe, especially within the conformity assessment bodies for reviewing the CE certification applications. As suggested by the FDA14, there is a great need for more harmonization in the AI domain, more specifically of the development of Good Machine Learning Practice (GMLP) through the creation of consensus standards and other guidelines.
References
1. Van Laere S, Muylle KM, Cornu P. Clinical Decision Support and New Regulatory Frameworks for Medical Devices: Are We Ready for It? – A Viewpoint Paper. IJHPM 2021. Available at https://www.ijhpm.com/?_action=article&au=54346&_au=Van%20Laere,%20Sven
2. U.S. Government Publishing Office. Public Law 114-255 – 114th Congress (in short: 21st Century Cures Act). Available at https://www.govinfo.gov/app/details/ PLAW-114publ255. Published 2016.
3. European Parliament and Council. Regulation (EU) 2017/745 (in short: Medical Device Regulation (MDR)). Available at https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02017R0745-201705052017 . Published 2017.
4. US Food and Drug Administration. Policy for Device Software functions and Mobile Medical Application. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-device-software-functions-and-mobile-medical-applications . Published 2019.
5. US Food and Drug Administration. Draft guidance on Clinical Decision Support (CDS) Software. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-decision-support-software . Published 2019
6. US Food and Drug Administration. Changes to existing Medical Software. Available at Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act – Draft Guidance for Industry and Food and Drug Administration Staff (fda.gov). Published 2019.
7. US Food and Drug Administration. General Wellness: Policy for Low Risk devices. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-wellness-policy-low-risk-devices . Published 2019.
8. US Food and Drug Administration. Off-the Shelf Software Use in Medical Devices Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/shelf-software-use-medical-devices . Published in 2019.
9. US Food and Drug Administration. Medical Device Data Systems, Medical Image Storage Devices, and Medical Image Communications Devices. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/medical-device-data-systems-medical-image-storage-devices-and-medical-image-communications-devices . Published in 2019.
10. Medical Device Coordination Group Document MDCG 2019-11. Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU)
2017/746 – IVDR. Available at https://ec.europa.eu/docsroom/documents/37581 . Published October 2019.
11. Global Harmonization Task Force. GHTF/SG1/N071:2012. Definition of the Terms ‘Medical Device’ and ‘In Vitro Diagnostic (IVD) Medical Device’. Available at https://www.imdrf.org/documents/ghtf-final-documents/ghtf-study-group-1-pre-market-evaluation . Published in 2012.
12. US Food and Drug Administration Safety and Innovation Act (FDASIA) Health IT Report, April 2014, available at https://www.fda.gov/about-fda/cdrh-reports/fdasia-health-it-report
13. IMDRF SaMD Working Group. “Software as a Medical Device”: Possible Framework for Risk Categorization and Corresponding Considerations. http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-140918-samd-framework-risk-categorization-141013.pdf . Published 2014.
14. US Food and Drug Administration. Artificial Intelligence/Machine Learning (AI/ML) based software as a medical device (SaMD) Action Plan. Available at https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-software-medical-device. Published January 2021.
Please find the complete paper here.
Topics: #healthcare #lifeSciences #medicaldevices #medtech #medicaltechnology #MedSysCon #FDA #ClinicalDecisionSupportSoftware #RegulatoryFramework
For further information please get in touch with us:
+49-176-57694801