
13 Feb
ISO 14971:2019 — Clarifying Benefit, Risk, & Benefit-Risk
Edwin L. Bills, member, ISO TC 210 JWG, wrote in the Guest Column on Med Device Online from February 8, 2021: “Discussion of the term “benefit” first appeared in the first edition of ISO 14971, which appeared in 2000. The requirement of controlling risk allowed the use of “benefit” as an alternative method of releasing a product when a residual risk was evaluated as “unacceptable.” Residual risk is that risk remaining after risk controls are applied, so, in this case, all possible risk controls were applied, and the residual risk still could not reach acceptable levels. The product should not be put on the market if the benefit does not outweigh the risk in a documented benefit-risk analysis. It would be up to regulators to ask the question, but you should not move product forward if the benefit does not outweigh the risk or until further risk reduction could be made.
One of the dramatic examples of the use of risk-benefit evaluation was the original mechanical heart, which had an associated mortality rate of 40% when it was used. This risk was considered unacceptable until measured against the benefit. The benefit was that the mortality rate was 70% without the use of the device. Clearly, the improved benefit outweighed the risk, and the device was ultimately cleared for use by FDA. However — and here is a consideration that must be part of postmarket review (see previous production and post-production article) — in less than a year, what was considered “state of the art” (another ISO 14971:2019 defined term in 3.28) changed and a competing product was released with an associated 30% mortality rate. Now, the original product was no longer acceptable — not because it was defective or the risk changed, but because the state of the art changed and a safer product was available. The original product had to be withdrawn until changes could be made to reach the new state of the art.
Note that the term “risk-benefit” was used in the 2000 edition and the 2007 edition of the standard. In the 2019 edition of the standard, you will find the term is now “benefit-risk” because regulators felt that manufacturers were only looking at benefit as an afterthought and thus wanted to emphasize benefit first instead of risk. Nowhere in regulation or standards will you find this term defined; however, you can find definition of “risk” in ISO 14971 (now at 3.18 in the 2019 version) going back to the original edition. In the standard, the definition is and has been “combination of the probability of occurrence of harm and the severity of that harm.” This definition was adopted by the regulators and appears in the EU’s Medical Device Regulation (MDR) and In Vitro Diagnostics Regulation (IVDR).
The FDA Quality System Regulation (QSR), 21 CFR 820, appeared several years before ISO 14971 and did not have “risk” as a defined term in the regulation. However, FDA was aware that the state of the art was changing. In the Preamble to the QSR in Comment 83, FDA indicated it was participating in ISO Technical Committee 210, working in the area of medical device risk. The FDA is currently considering updating its regulation to align with ISO 13485, in which the term “risk” carries the ISO 14971 definition.
In Essential Principles of Safety and Performance of Medical Devices and IVD Medical Devices, IMDRF/GRRP WG/N47 FINAL:2018, the International Medical Device Regulators Forum (IMDRF) uses the same “risk” definition as ISO 14971 in 3.35.
So far, we have half the equation, “risk,” well defined. The term “benefit,” however, had been undefined by regulators or in standards until ISO 14971:2019. Here, a couple of things happened:
- “Benefit” is now defined, in 3.2, as “positive impact or desirable outcome of the use of a medical device on the health of an individual, or positive impact on patient management or public health.” A Note 1 to Entry expands information to help clarify the implications of “benefit.”
- Since then, regulators, including FDA (see the four guidance documents in Refs. 1–4) and the European Commission (EC), adopted the term “benefit-risk” instead of “risk-benefit” as was previously used in the standard. ISO TC 210 JWG1, the authors of the risk management standard (ISO 14971) and technical report (ISO TR 24971), updated the standard and technical report to the new term for the new releases in 2019 and 2020, respectively.
We now have defined terms updated to current regulatory thinking, in the ISO 14971 standard and ISO TR 24971 technical report, so we now understand what we are talking about. Of course, we defined “risk” and “benefit” individually, and not as a combined term, but we have some understanding of the terms.
The 2019 edition of the standard indicates in 7.4 Benefit-Risk Analysis that “benefit-risk analysis” may be used to address unacceptable individual residual risks. It refers to ISO TR 24971’s discussion (via guidance, not requirements) of some aspects and possible methods of this analysis. This section only deals with the individual risks, and the standard (7.4) only requires that individual unacceptable residual risks undergo this analysis. Some regulatory bodies may require that all individual risks, regardless of acceptability, undergo this analysis. This is not in conflict with the standard, so you must apply this regulatory requirement in addition to the requirement of the standard if you are serving a market where this additional requirement exists. How to perform these analyses is beyond the space allowance we have here, but the discussion referred to earlier in 24971 is more than we could successfully perform here.
For individual risks, then, the 7.4 requirement in the standard only addresses individual unacceptable risks, not all risks. Regulators may impose additional requirements. However, we need to go further in the standard to uncover additional requirements for “benefit-risk analysis.”
How To Evaluate Overall Residual Risk
In ISO 14971’s Clause 8, Evaluation of Overall Residual Risk, we find a universal requirement applicable to all devices, regardless of risk acceptability. The standard requires, “the manufacturer shall evaluate the overall residual risk posed by the medical device, taking into account the contributions of all residual risks, in relation to the benefits of the intended use…” (emphasis mine). This means for every device, an overall residual risk evaluation shall consider the benefit compared to the risk of using the device. The benefit should outweigh the risk. Fortunately, ISO TR 24971:2020 includes 2.5 pages of guidance on methods you can use to perform this analysis. The technical report in 8.3 a)–f) provide some possible approaches, indicating there is no preferred approach. The evaluators should be experienced in the use of the device and the environment of use, which are both critical aspects of such an evaluation. One of the key tools I have used in this type of risk evaluation is a visual tool presenting all of the risks of the device simultaneously on a risk chart so that it may be easier to understand the device’s risk profile in such an analysis. ISO TR 24971 8.3 b) explains this approach to using visual representations of residual risks to understand where problems may exist in the overall residual risk of the device and where efforts can be applied to give the best results on reducing residual risk.
ISO TR 24971’s Clause 8.1 discusses that there is no preferable way to perform overall residual risk analysis, and that judgment is involved. As a result, it is essential that professionals with knowledge, experience, and authority (including application specialists and clinical specialists or users) perform this evaluation. In my experience, a team that includes medical professionals current with device experience, product liability insurance experts, experienced design members, and risk management experts not associated with the project would provide the needed expertise to perform this evaluation. Adequate preparation, including review of the entire risk management file for the device prior to conducting the overall residual risk evaluation and review of documentation of comparable devices, is necessary and may provide valuable information. Additionally, the overall residual risk acceptability criteria provided for the device might be different from the individual residual risk acceptability criteria. This criteria must be included in the risk management plan for the device prior to conducting the evaluation. ISO TR 24971 Clause 8.2 provides information on the inputs for the evaluation.
An important aspect of the requirements in Clause 8 is the requirement that “the manufacturer shall inform the user of significant residual risks, and shall include the necessary information in the accompanying documentation.” A limited discussion of the rationale about what is significant for this disclosure appears in ISO 14971 Annex A.2.8.
It is important to understand that only Clauses 1–10 of ISO 14971:2019 contain requirements. All other parts of the standard and all parts of ISO TR 24971 are only guidance to provide aid in constructing your risk management system. Those sections are not requirements. This misunderstanding has caused a great deal of problems for those implementing the standard in the past.
That covers the risk side. Now for the benefit side of the equation.
How To Evaluate Benefit
While engineers love numbers and want to assign values to such an analysis, it is not possible to do this numerically for benefit, because it consists of many independent factors. This effort should involve medical/clinical risk management team members, as they are more qualified to perform benefit analysis. Additionally, the evaluation team must consider any alternative treatment modalities, including pharmaceuticals and biologics, when considering benefit-risk. If the alternative treatments provide the benefit at a lower risk, then the necessary benefit-risk may not be achieved to place the device on the market. ISO TR 24971:2020’s Clause 7.4 discusses benefit-risk analysis, with some additional information is provided in Clause 8.3 a) and c).
First, for a more complete understanding of benefit, the definition of the term in ISO 14971:2019 Clause 3.2 is essential:
benefit
positive impact or desirable outcome of the use of a medical device on the health of an individual, or a positive impact on patient management or public health
Note 1 to entry: Benefits can include positive impact on clinical outcome, the patient’s quality of life, outcomes related to diagnosis, positive impact from diagnostic devices on clinical outcomes, or positive impact on public health.
ISO 24971:2020 7.4.2 mentions factors necessary to consider when evaluating benefit, such as:
- Expected device performance
- Expected clinical outcome at that performance level
- Competing devices
- Alternative treatment modalities.
Confidence of the benefit estimate is important and is based on reliability of information. One problem is trying to compare different outcomes (pain versus mobility) and short-term versus long-term effects. Other considerations include:
- Type of benefit
- Magnitude of benefit
- Probability the patient will experience the benefit
- Duration of the benefit.
Availability of clinical data to support benefit determinations impacts benefit estimates (remember, data prior to getting real data from users after release is just an estimate and has associated uncertainty). ISO TR 24971:2020’s Clause 7.4.5 has three examples of benefit-risk analysis; although they are provided for individual benefit-risk, they can be extrapolated to the necessary overall benefit-risk requirement in Clause 8.
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

12 Dec
ISO TR 249712020 Brings Clarity To Risk Acceptability In ISO 14971
Edwin L. Bills, member, ISO TC 210 JWG, wrote in the Guest Column on Med Device Online: “Periodically, standards are revisited by international and national committees to determine if they are still current or need revision or withdrawal. Through the voting process and based on comments received during the voting period in early 2016 on both ISO 14971:2007 (the standard for application of risk management to medical devices) and ISO TR 24971:2013 (the guidance on the application of ISO 14971), ISO determined that these documents needed to be updated and the policy for risk acceptability should be revised for clarity.
Among the issues that needed attention was the requirement for top management responsibility for establishing a policy for determining risk acceptability criteria in clause 3.2 of ISO 14971:2007, which was addressed in the ISO 14971:2019 Third Edition. The policy for risk acceptability criteria has been a part of ISO 14971 since its inception in 2000, but many have raised questions about what this requirement means. Some have created risk matrices or risk charts to describe the requirement, but that is not a correct interpretation of the standard, and should not be used.
The resulting revision of ISO TR 24971:2013 — ISO TR 24971:2020 — provides extensive guidance in the informative annexes, discussions of the requirements in ISO 14971:2019, and further discussion of the terms “benefit” and “benefit-risk analysis.” It does not add any requirements. It is only guidance or help for those implementing the standard.
The Requirement
ISO 14971:2019 clause 4.2 requires that:
Top management shall define and document a policy for establishing criteria for risk acceptability. This policy shall provide a framework that ensures that criteria for risk acceptability are based on applicable national/regional regulations, relevant international standards, take into account the generally acknowledged state of the art, and known stakeholder concerns.
An additional note, which is informative and is not a requirement, provides more information regarding the approach to risk control in the risk acceptability policy:
NOTE 1 The manufacturer’s policy for establishing criteria for risk acceptability can define the approaches to risk control: reducing risk as low as reasonably practicable, reducing risk as low as reasonably achievable, or reducing risk as far as possible without adversely affecting the benefit-risk ratio. See ISO/TR 24971[9] for guidance on defining such policy.
These are the same requirements as in ISO 14971:2007 clause 3.2, and also clause 3.3(a) in the earlier 2000 edition. Unfortunately, these requirements were not further explained until ISO TR 24971:2013 was released. Even then, more information with examples might have helped in understanding this requirement, and that is what ISO TR 24971:2020 Annex C provides.
Past Incorrect Implementations
Because of a lack of direction from the technical committee in the earlier editions of the standard and the technical report, many interpretations of the requirement came into being. Among them was interpretation defining the acceptable risk as a risk matrix using a two-dimensional chart with one axis being identified as probability of occurrence of harm and the other axis being severity of harm with appropriate levels being chosen by the manufacturer and identified by notations along with the risk chart. Many could not agree on a standard appearance of such a matrix and it seemed every interpretation was different with the zero points being any of the four corners, and exchanging the severity and probability axes, though most often the zero point was the lower left or upper left corner. A line through the matrix was identified by the company as identifying the boundary between acceptability and unacceptability. This was most often, though not always, shown as the boundary between the Intolerable Risks and the Investigate for Further Risk Reduction regions, though other terms were used to identify the regions.
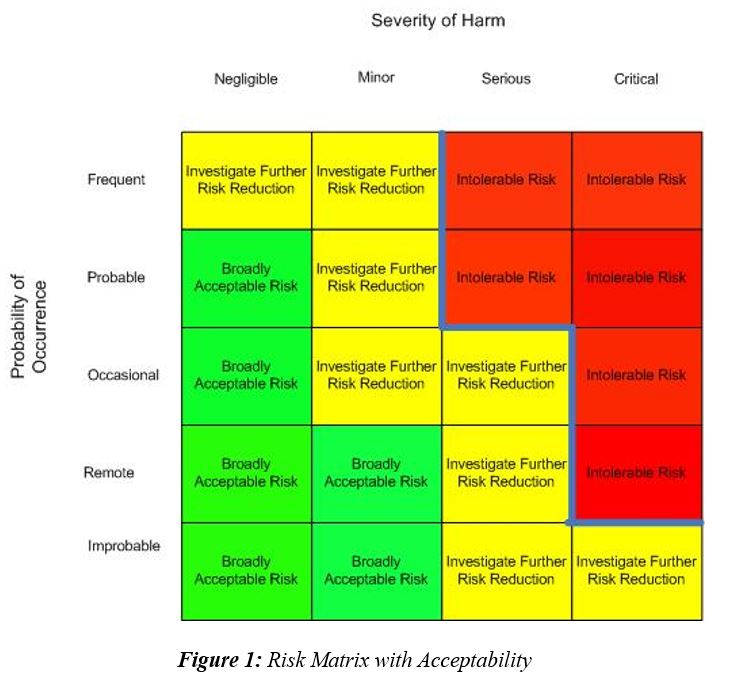
Some incorrectly used a technique to establish risk acceptability from earlier editions of failure modes and effects analysis (FMEA) identified as risk priority number (RPN). It is important to note the RPN technique was removed from the automotive industry FMEA as it was very confusing and inaccurate, and led to incorrect choices. It should not be used by the medical device industry to establish risk acceptability for much the same reasons. It is not establishing risk acceptability according to any of the risk acceptability approaches recognized in medical device risk management.
Instead of having Acceptable Risk and Unacceptable Risk regions defined by policy, some have added an intermediate region erroneously identified as As Low As Reasonably Practicable (ALARP). ALARP is not a region on a chart but is an approach for identifying the process of how far to reduce risk. Many companies continued to use this inaccurate interpretation at least until the release of the ISO 14971:2019 standard. The middle region is more correctly an Investigate Further Risk Control region, (ISO TR 24971:2020 Figure C1) meaning that risks that fall on a risk chart in this region should be further reduced by applying additional risk control measures.
In the European Union (EU), the confusion on acceptable versus unacceptable risks was muddied in the release of the EN ISO 14971:2012 edition, which indicated a company could not use the ALARP approach but should reduce risk using the As Far As Possible (AFAP) approach following the three medical device directives in Europe. The EN 2012 standard did not identify a process for identifying how the level required could be accomplished, causing more confusion. Providing objective evidence to auditors and regulators that AFAP has been reached is difficult, if not impossible. One more risk control could always be applied with some degree of improvement, even if it is infinitesimal. Making the decision of how much improvement is enough is difficult. The EN ISO 14971:2012 version was withdrawn by CEN with the release of EN ISO 14971:2019.
New Regulations
The recent Medical Device Regulations (MDR) and In Vitro Diagnostics Regulations (IVDR) regulations replacing the directives in the EU has not improved the situation to any great degree, requiring the manufacturer to reduce risks AFAP without impacting the benefit-to-risk ratio, yet the two regulations do not identify what a benefit-to-risk ratio is or how to accomplish reaching the AFAP goal. The term “benefit” is defined in ISO 14971:2019 3.2, but nowhere else in regulations, guidance, or standards. An extensive discussion, with examples, of “benefit” and “benefit-risk analysis” is found in ISO TR 24971:2020 7.4. This approach seems to indicate that risk charts may not be useful, as each risk must be reduced to AFAP on its own without consideration of acceptable risks.
ISO TR 24971:2020 Clarifications
A clarification in Annex C of ISO TR 24971:2020 indicates that individual risks may have different levels of risk acceptability than the overall residual risk. If a device has these different levels for the two types of risk — individual and overall residual — then these differing levels must be identified in the product risk management plan.
In addition, Annex C identifies five possible elements of the policy for risk acceptability criteria then provides a possible example for each of these elements:
- Purpose
- Scope
- Factors and considerations for determining risk acceptability criteria
- Approaches to risk control (e.g., ALARP, AFAP, As Low As Reasonably Achievable [ALARA], As Low As Possible [ALAP])
- Requirements for review and approval
The section requiring the most work to develop in the criteria for risk acceptability is the section on factors and considerations. It will require some effort to identify the appropriate elements for this section. There is guidance in Annex C to assist in this element.
“Approaches” is a decision point for the company management in selecting the appropriate approach. This decision may be influenced by the regulatory requirements, such as the EU’s use of AFAP (without impacting the benefit-to-risk ratio) in the MDR and IVDR.
To complete the risk acceptability criteria process, ISO TR 24971:2020 includes an additional set of examples comparing the elements of a policy, the acceptability criteria, and the evaluation of the results for the four elements in a policy:
- Regulatory requirements for the intended markets can be found and applied to the device risk acceptability criteria, but if the markets change, it is important to update the risk acceptability criteria with new market requirements.
- International standards that impact a product from the product-specific to the cross-cutting horizontal product safety standards must be considered in the development of the risk acceptability criteria; this might include electrical safety standards such as IEC 60601-1 and its family of standards.
- State of the art, which is a concept easily confused.*
- Stakeholder concerns can be collected from focus groups and product experts in the use of their particular product type in the environment of the intended use for their product type. It is important here to use the input from experts in the current use of the product based on current medical practice.”
Please find the complete article here.
For further information please get in touch with us:
+49-176-57694801

23 Feb
FDA recognizes ISO 14971:2019 as consensus standard
On Jan 14, 2020 the US FDA has granted Recognized Consensus Standard status to the third edition of the ISO 14971 risk management standard for medical devices and IVD products ( see: FDA Recognized Consensus Standards ).
Transition Period
The transition period of the previous standard edition, ISO 1497:2007, will last through late 2022. Therefore, any declarations of conformity to ISO 14971:2007 included in medical device and IVD premarket submissions will be accepted by the FDA until December 25, 2022.
Afterwards a medical device submission requires a declaration of conformity to the latest edition ISO 14971:2019.
Please get in touch with us for any further support:
+49-176-57694801